Imatinib
Description
Evolution of Targeted Therapies and Tyrosine Kinase Inhibitors
The development of targeted therapies was driven by a deeper understanding of cancer biology, specifically the role of aberrant signaling pathways in oncogenesis. Tyrosine kinases, a family of enzymes responsible for adding phosphate groups to tyrosine residues on proteins (phosphorylation), play crucial roles in cellular processes such as growth, division, and communication mdpi.comwikipedia.orgoncodesign-services.comnih.govresearchgate.net. When these kinases become dysregulated, often through mutations or overexpression, they can drive uncontrolled cell proliferation and survival, contributing to cancer development mdpi.comnih.govresearchgate.net.
The concept of selectively inhibiting tyrosine phosphorylation dates back to seminal research in 1988, which described compounds capable of inhibiting the catalytic activity of the epidermal growth factor receptor (EGFR) wikipedia.org. This early work laid the groundwork for the systematic search and discovery of small-molecule inhibitors that could specifically target these enzymatic activities. The subsequent decades witnessed the evolution of TKIs, moving from broader-acting agents to more selective compounds designed to overcome resistance mechanisms that often emerge during treatment mdpi.comnih.govamegroups.org. This continuous refinement in TKI design has significantly improved therapeutic efficacy and patient outcomes mdpi.com.
Imatinib as a Prototypical Tyrosine Kinase Inhibitor in Research Contexts
This compound, also known by its research designation STI-571, stands as a quintessential example and a prototype among tyrosine kinase inhibitors, marking a watershed moment in targeted cancer therapy research nih.govcapes.gov.brnih.govjnccn.orgonclive.comscielo.br. Its introduction in 2001 revolutionized the treatment of chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST), diseases previously associated with poor prognoses onclive.comnih.govnih.govecancer.org.
This compound is a 2-phenyl amino pyrimidine derivative and functions as a specific inhibitor of several key tyrosine kinases, including BCR-ABL, c-KIT, and platelet-derived growth factor receptor (PDGFR) nih.govwikipedia.orgdroracle.aimrc.ac.uk. Its mechanism of action involves competitively binding to the adenosine triphosphate (ATP) binding site of these kinases, effectively locking them in an inactive, closed, or self-inhibited conformation wikipedia.orgdroracle.aiashpublications.orgresearchgate.netresearchgate.net. This binding prevents the transfer of phosphate groups from ATP to tyrosine residues on substrate proteins, thereby inhibiting downstream signaling pathways essential for cancer cell growth and survival, ultimately leading to apoptosis and reduced proliferation in affected cells wikipedia.orgdroracle.aiashpublications.orgresearchgate.netresearchgate.net.
The groundbreaking research surrounding this compound has provided detailed insights into its effectiveness. In chronic myelogenous leukemia, research demonstrated that this compound significantly improved patient outcomes. The landmark IRIS (International Randomized Interferon versus STI571) study, for instance, reported complete hematological responses in 95.3% of patients and complete cytogenetic responses in 73.8% of patients with chronic phase CML nih.gov. Long-term follow-up studies further confirmed these remarkable results, with 98% of patients achieving complete hematological response and 87% achieving complete cytogenetic response after six years nih.gov. This success has led to a dramatic improvement in the life expectancy of CML patients, approaching that of the general population oncology-central.comnih.govonclive.comnih.gov.
Despite its profound success, research also identified that resistance to this compound can develop, often due to specific mutations within the BCR-ABL kinase domain that alter the drug's binding affinity nih.govnih.govwikipedia.orgresearchgate.netresearchgate.net. This understanding spurred further research and the subsequent development of second- and third-generation tyrosine kinase inhibitors designed to overcome these resistance mechanisms, showcasing this compound's foundational role in advancing targeted therapy research mdpi.comnih.govresearchgate.net.
The following table summarizes key research findings regarding this compound in CML and GIST:
| Disease | Key Research Finding | Relevant Kinase(s) |
| Chronic Myelogenous Leukemia (CML) | - Landmark IRIS study reported complete hematological response in 95.3% and complete cytogenetic response in 73.8% of patients in chronic phase CML. nih.gov- Six-year follow-up of IRIS trial showed 98% complete hematological response and 87% complete cytogenetic response. nih.gov- Patients' life expectancy on this compound has become comparable to the general population. oncology-central.comnih.govonclive.comnih.gov | BCR-ABL nih.govcapes.gov.brnih.govwikipedia.orgdroracle.aimrc.ac.ukresearchgate.net |
| Gastrointestinal Stromal Tumors (GIST) | - Demonstrated strong long-term survival, with a 10-year overall survival estimate of 23% for patients with advanced GIST in the SWOG S0033 trial. ecancer.org- Survival rates significantly higher for patients with KIT exon-11 mutant GISTs. ecancer.org | c-KIT, PDGFR nih.govcapes.gov.brnih.govwikipedia.orgdroracle.aimrc.ac.ukresearchgate.net |
Properties
IUPAC Name |
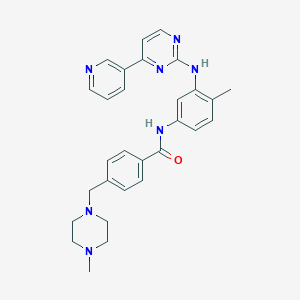
4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide |
Source


|
|---|---|---|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
InChI |
InChI=1S/C29H31N7O/c1-21-5-10-25(18-27(21)34-29-31-13-11-26(33-29)24-4-3-12-30-19-24)32-28(37)23-8-6-22(7-9-23)20-36-16-14-35(2)15-17-36/h3-13,18-19H,14-17,20H2,1-2H3,(H,32,37)(H,31,33,34) |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
InChI Key |
KTUFNOKKBVMGRW-UHFFFAOYSA-N |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Canonical SMILES |
CC1=C(C=C(C=C1)NC(=O)C2=CC=C(C=C2)CN3CCN(CC3)C)NC4=NC=CC(=N4)C5=CN=CC=C5 |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Molecular Formula |
C29H31N7O |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
DSSTOX Substance ID |
DTXSID3037125 |
Source
|
| Record name | Imatinib | |
| Source | EPA DSSTox | |
| URL | https://comptox.epa.gov/dashboard/DTXSID3037125 | |
| Description | DSSTox provides a high quality public chemistry resource for supporting improved predictive toxicology. | |
Molecular Weight |
493.6 g/mol |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Physical Description |
Solid |
Source
|
| Record name | Imatinib | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014757 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
Solubility |
Very soluble in water at pH < 5.5 (mesylate salt), 1.46e-02 g/L |
Source
|
| Record name | Imatinib | |
| Source | DrugBank | |
| URL | https://www.drugbank.ca/drugs/DB00619 | |
| Description | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. | |
| Explanation | Creative Common's Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/legalcode) | |
| Record name | Imatinib | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014757 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
CAS No. |
152459-95-5 |
Source
|
| Record name | Imatinib | |
| Source | CAS Common Chemistry | |
| URL | https://commonchemistry.cas.org/detail?cas_rn=152459-95-5 | |
| Description | CAS Common Chemistry is an open community resource for accessing chemical information. Nearly 500,000 chemical substances from CAS REGISTRY cover areas of community interest, including common and frequently regulated chemicals, and those relevant to high school and undergraduate chemistry classes. This chemical information, curated by our expert scientists, is provided in alignment with our mission as a division of the American Chemical Society. | |
| Explanation | The data from CAS Common Chemistry is provided under a CC-BY-NC 4.0 license, unless otherwise stated. | |
| Record name | Imatinib [INN:BAN] | |
| Source | ChemIDplus | |
| URL | https://pubchem.ncbi.nlm.nih.gov/substance/?source=chemidplus&sourceid=0152459955 | |
| Description | ChemIDplus is a free, web search system that provides access to the structure and nomenclature authority files used for the identification of chemical substances cited in National Library of Medicine (NLM) databases, including the TOXNET system. | |
| Record name | Imatinib | |
| Source | DrugBank | |
| URL | https://www.drugbank.ca/drugs/DB00619 | |
| Description | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. | |
| Explanation | Creative Common's Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/legalcode) | |
| Record name | Imatinib | |
| Source | DTP/NCI | |
| URL | https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&outputformat=html&searchlist=759854 | |
| Description | The NCI Development Therapeutics Program (DTP) provides services and resources to the academic and private-sector research communities worldwide to facilitate the discovery and development of new cancer therapeutic agents. | |
| Explanation | Unless otherwise indicated, all text within NCI products is free of copyright and may be reused without our permission. Credit the National Cancer Institute as the source. | |
| Record name | Imatinib | |
| Source | DTP/NCI | |
| URL | https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&outputformat=html&searchlist=743414 | |
| Description | The NCI Development Therapeutics Program (DTP) provides services and resources to the academic and private-sector research communities worldwide to facilitate the discovery and development of new cancer therapeutic agents. | |
| Explanation | Unless otherwise indicated, all text within NCI products is free of copyright and may be reused without our permission. Credit the National Cancer Institute as the source. | |
| Record name | Imatinib | |
| Source | EPA DSSTox | |
| URL | https://comptox.epa.gov/dashboard/DTXSID3037125 | |
| Description | DSSTox provides a high quality public chemistry resource for supporting improved predictive toxicology. | |
| Record name | IMATINIB | |
| Source | FDA Global Substance Registration System (GSRS) | |
| URL | https://gsrs.ncats.nih.gov/ginas/app/beta/substances/BKJ8M8G5HI | |
| Description | The FDA Global Substance Registration System (GSRS) enables the efficient and accurate exchange of information on what substances are in regulated products. Instead of relying on names, which vary across regulatory domains, countries, and regions, the GSRS knowledge base makes it possible for substances to be defined by standardized, scientific descriptions. | |
| Explanation | Unless otherwise noted, the contents of the FDA website (www.fda.gov), both text and graphics, are not copyrighted. They are in the public domain and may be republished, reprinted and otherwise used freely by anyone without the need to obtain permission from FDA. Credit to the U.S. Food and Drug Administration as the source is appreciated but not required. | |
| Record name | Imatinib | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014757 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
Melting Point |
226 °C (mesylate salt) |
Source
|
| Record name | Imatinib | |
| Source | DrugBank | |
| URL | https://www.drugbank.ca/drugs/DB00619 | |
| Description | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. | |
| Explanation | Creative Common's Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/legalcode) | |
| Record name | Imatinib | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014757 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
Molecular and Cellular Mechanisms of Imatinib Action
Tyrosine Kinase Target Specificity and Inhibition
Imatinib primarily functions by inhibiting the activity of specific tyrosine kinases, including the BCR-ABL oncoprotein, platelet-derived growth factor receptor (PDGFR) kinase, and c-KIT kinase. This selective inhibition is achieved through competitive binding at the ATP-binding site of these kinases, preventing them from phosphorylating their substrate proteins and halting downstream signaling pathways essential for cell growth and survival. drugbank.comdroracle.aiaacrjournals.org this compound exhibits a high level of selectivity, with its activity against these protein kinases being at least 100-fold lower than for a large number of other tyrosine and serine/threonine kinases. nih.gov
BCR-ABL Oncoprotein Kinase Inhibition
The BCR-ABL fusion protein is a constitutively active tyrosine kinase resulting from the Philadelphia chromosome abnormality (t(9;22) translocation) in chronic myeloid leukemia (CML). drugbank.compharmgkb.org This aberrant oncoprotein drives uncontrolled cell proliferation and survival in CML. drugbank.comnih.govdroracle.aipharmgkb.org this compound specifically targets and inhibits the BCR-ABL tyrosine kinase by binding to its ATP-binding pocket in the inactive conformation. drugbank.comnih.govresearchgate.nettandfonline.comresearchgate.net This binding prevents the transfer of a phosphate group from ATP to tyrosine residues on substrate proteins, thereby blocking their activation and the subsequent transmission of proliferative signals to the nucleus. nih.govresearchgate.netresearchgate.net Consequently, this compound effectively suppresses the growth of BCR-ABL-positive cells and induces apoptosis, while largely sparing normal cells. nih.govpharmgkb.org
Platelet-Derived Growth Factor Receptor (PDGFR) Kinase Inhibition
This compound also acts as a potent inhibitor of the platelet-derived growth factor receptor (PDGFR) kinase, including both alpha and beta isoforms. drugbank.comdroracle.aicellagentech.com PDGFRs are receptor tyrosine kinases that regulate various cellular processes such as proliferation, differentiation, migration, and survival upon binding to their ligand, platelet-derived growth factor (PDGF). scbt.com In certain cancers, such as gastrointestinal stromal tumors (GISTs) and some myeloproliferative disorders, mutations in PDGFR can lead to its constitutive activation, driving aberrant cell growth. droracle.airesearchgate.netaacrjournals.orgonclive.com this compound inhibits PDGFR activity by binding selectively to its ATP-binding site, stabilizing the inactive conformation of the receptor. scbt.comresearchgate.net This interaction prevents autophosphorylation and the phosphorylation of downstream substrate molecules, thereby inhibiting downstream signaling cascades and subsequent cellular proliferation and survival. scbt.comresearchgate.netaacrjournals.org
c-KIT Kinase Inhibition
The c-KIT receptor tyrosine kinase, also known as the stem cell factor receptor (SCF-R), plays a crucial role in normal cell growth and development. drugbank.comdroracle.ai However, activating mutations in c-KIT are frequently observed in gastrointestinal stromal tumors (GISTs), leading to constitutive kinase activity and uncontrolled cell growth. drugbank.compharmgkb.orgaacrjournals.orgresearchgate.net this compound effectively inhibits both wild-type and certain mutant forms of c-KIT by occupying the ATP-binding pocket of the kinase domain in its inactive conformation. drugbank.comaacrjournals.orgcellagentech.comresearchgate.netpnas.org This binding prevents the phosphorylation of c-KIT substrates and subsequent activation of downstream signaling pathways, including the MAPK and Akt pathways, which are critical for cell proliferation and survival. cellagentech.comresearchgate.netstanford.edu It's important to note that certain c-KIT mutations, such as D816V in systemic mastocytosis, can render the kinase constitutively active in a conformation that this compound does not effectively inhibit, leading to resistance. pnas.orgwikipedia.orgashpublications.org
Molecular Interactions at the ATP-Binding Pocket
This compound's inhibitory mechanism involves its specific binding to the ATP-binding pocket within the kinase domain of its target enzymes (BCR-ABL, PDGFR, c-KIT). drugbank.comnih.govdroracle.airesearchgate.netresearchgate.netwikipedia.org This pocket is typically occupied by adenosine triphosphate (ATP) during the normal phosphorylation cycle. This compound acts as a competitive inhibitor, binding to this site and preventing ATP from accessing it. researchgate.netresearchgate.net this compound generally binds to the inactive conformation of the kinase. nih.govtandfonline.comscbt.comnih.gov For example, in BCR-ABL, this compound binding locks the kinase domain in an inactive, DFG-out/AL-closed conformation, preventing substrate binding and phosphorylation. nih.gov Molecular dynamics simulations indicate that this compound interacts with various regions of the Abl kinase, including the ATP-binding site, the interface with the SH2 domain, the myristoyl-binding site, the αG helix region of the C-lobe, and the PIF-binding site of the N-lobe. nih.gov At physiological pH, the 1-methylpiperazin-1-ium group nitrogen of this compound may be protonated, and simulations suggest that this compound binds to Abl in its protonated, positively-charged form. researchgate.net
Prevention of Substrate Phosphorylation
The core consequence of this compound binding to the ATP-binding pocket of its target kinases is the prevention of substrate phosphorylation. nih.govresearchgate.netresearchgate.netwikipedia.org By occupying the ATP-binding site, this compound blocks the transfer of a phosphate group from ATP to the tyrosine residues of target proteins. nih.govdroracle.airesearchgate.netresearchgate.netresearchgate.net This inhibition means that the normal substrates of these kinases cannot be phosphorylated and thus cannot become activated. As a result, the downstream signaling cascades that depend on these phosphorylation events are interrupted, leading to the inhibition of cell proliferation and the induction of apoptosis in affected cancer cells. nih.govdroracle.aiaacrjournals.orgresearchgate.netresearchgate.net
Downstream Signaling Pathway Modulation
The inhibition of key tyrosine kinases by this compound leads to the modulation of several downstream signaling pathways that are crucial for cell survival, proliferation, and differentiation. For instance, the BCR-ABL pathway controls numerous downstream pathways implicated in neoplastic growth, including the Ras/MAPK pathway (cellular proliferation), Src/Pax/Fak/Rac pathway (cellular motility), and PI/PI3K/AKT/BCL-2 pathway (apoptosis pathway). drugbank.com By blocking BCR-ABL, this compound disrupts these pathways, leading to inhibited proliferation and induced apoptosis of leukemic cells. nih.gov
In the context of PDGFR and c-KIT inhibition, this compound prevents the activation of pathways such as the MAPK/ERK pathway and the PI3K/Akt pathway, both of which are critical for cell growth and survival. cellagentech.comaacrjournals.orgstanford.edu Research has shown that this compound can inhibit PDGFR-mediated signaling in fibroblasts and c-KIT activation in mast cells, leading to a reduction in the phosphorylation of downstream Akt and ERK. stanford.edu Furthermore, this compound's inhibition of BCR-ABL has been shown to prevent the tyrosine phosphorylation of Smad4, thereby restoring growth-suppressive TGF-β signaling, which is often disrupted in cancer cells. nih.gov This comprehensive modulation of downstream signaling pathways is central to this compound's therapeutic efficacy in various malignancies.
Ras/MapK Pathway Perturbations
The BCR-ABL oncogenic pathway exerts control over numerous downstream signaling cascades vital for neoplastic growth, including the Ras/MapK (Mitogen-activated protein kinase) pathway, which is intimately involved in cellular proliferation drugbank.com. This compound's primary inhibitory action on the BCR-ABL tyrosine kinase effectively disrupts this upstream activation, leading to downstream perturbations in the Ras/MapK pathway drugbank.comdroracle.ainih.gov. Research findings indicate that this compound can inhibit cell proliferation, partly through the inactivation of extracellular signal-regulated kinase (ERK), a key component of the MAPK pathway longdom.org. This suggests that this compound's therapeutic effects involve the dampening of hyperactive proliferative signals transmitted via the Ras/MapK cascade in malignant cells longdom.org.
Src/Pax/Fak/Rac Pathway Modulation
Another critical pathway regulated by the BCR-ABL pathway, and consequently modulated by this compound, is the Src/Pax/Fak/Rac pathway, which plays a significant role in cellular motility and adhesion drugbank.com.
Src Kinase : Src family kinases (SFKs) are non-receptor tyrosine kinases implicated in various signal transductions and are often aberrantly expressed or activated in numerous cancers researchgate.net. As BCR-ABL can activate SFKs, this compound's inhibition of BCR-ABL indirectly impacts Src activity drugbank.comnih.govresearchgate.net.
Focal Adhesion Kinase (FAK) : FAK is a tyrosine kinase crucial for tumor progression, affecting proliferation, survival, invasion, and metastasis aacrjournals.org. FAK activation typically involves autophosphorylation at tyrosine 397 (Y397), which then serves as a docking site for Src-family kinases and phosphatidylinositol 3-kinase (PI3K) aacrjournals.org. By modulating upstream kinases, this compound can influence FAK activity, thereby affecting cellular adhesion and motility drugbank.com. Experimental FAK inhibitors, such as GSK2256098 and PF-562271, have been developed to investigate the anti-tumor effects of FAK inhibition guidetopharmacology.orgnih.gov.
Rac : Rac1, a member of the Rho family of small GTPases, is involved in cytoskeletal reorganization, cell growth, and cell motility drugbank.comgenecards.org. Inhibition of the BCR-ABL pathway by this compound can disrupt the signaling that drives Rac activity, thereby impacting processes like cell migration and adhesion in malignant cells drugbank.com.
PI3K/AKT/BCL-2 Pathway Regulation
The PI3K/AKT/BCL-2 pathway is a major signaling cascade that regulates cell survival, proliferation, and growth, often contributing to oncogenic transformation when activated drugbank.commdpi.com. This compound's inhibitory effects on the BCR-ABL tyrosine kinase extend to the regulation of this pathway drugbank.com.
PI3K/AKT : this compound's inhibition of BCR-ABL can lead to the disruption of downstream PI3K activation, thereby blocking AKT signaling pathways longdom.orgjcpjournal.org. The activation of PI3K subsequently phosphorylates AKT, promoting cell survival mdpi.com. By inhibiting this axis, this compound contributes to the suppression of malignant cell growth jcpjournal.org.
BCL-2 : BCL-2 is a key anti-apoptotic protein nih.gov. The PI3K/AKT pathway's downstream effects include the regulation of BCL-2 family proteins. By downregulating survival signals transmitted through the PI3K/AKT pathway, this compound promotes an environment conducive to apoptosis by modulating the balance of pro- and anti-apoptotic BCL-2 family proteins drugbank.comnih.gov.
Cell Cycle Arrest Induction
This compound's molecular actions culminate in the induction of cell cycle arrest in various malignant cell types, thereby halting uncontrolled proliferation nih.gov. This effect is often observed in a dose-dependent manner longdom.orgplos.org. For instance, this compound treatment has been shown to induce G0/G1 phase arrest in human skin squamous cell carcinoma (SSCC) cells (A431), multiple myeloma cells, HepG2 hepatocellular carcinoma cells, and several osteosarcoma cell lines (MG-63, MOS-J, OSRGA) longdom.orgjcpjournal.orgplos.org. In other cell lines, such as HOS and POS-1 osteosarcoma cells, as well as MCF7 and T-47D breast cancer cells, this compound can lead to arrest in the S or G2/M phase plos.orgdovepress.com. Gastric cancer cells treated with this compound have also shown G2/M phase arrest spandidos-publications.com. Mechanistically, this cell cycle arrest is linked to the downregulation of S-phase kinase-associated protein 2 (SKP2) and the concomitant upregulation of P21CIP1 (p21) longdom.orgjcpjournal.org. SKP2 is an E3 ubiquitin ligase component that promotes the degradation of cell cycle inhibitors like P21CIP1, so its downregulation by this compound leads to P21CIP1 stabilization and cell cycle inhibition longdom.orgjcpjournal.org.
Table 1: Examples of this compound-Induced Cell Cycle Arrest in Various Cell Lines
| Cell Line/Type | Primary Cell Cycle Phase Arrest | Mechanism/Notes | Source |
| A431 SSCC cells | G0/G1 | Downregulation of SKP2, upregulation of P21CIP1 | longdom.org |
| Multiple Myeloma cells | G0/G1 | Increased P21CIP1 protein abundance | longdom.org |
| HepG2 Hepatocarcinoma cells | G0/G1 | Inhibition of SKP2, upregulation of p21 and p27 | jcpjournal.org |
| MG-63, MOS-J, OSRGA Osteosarcoma cells | G0/G1 | Dose-dependent, anti-proliferative effect | plos.org |
| HOS, POS-1 Osteosarcoma cells | S | Dose-dependent, anti-proliferative effect | plos.org |
| MCF7, T-47D Breast Cancer cells | G2/M | Significant cytostatic increase with reduction in S phase | dovepress.com |
| Gastric Cancer cells (AGS) | G2/M and sub-G1 | Time- and dose-dependent increase in cells in sub-G1 and G2/M phases | spandidos-publications.com |
Apoptosis Induction in Malignant Cells
Beyond cell cycle arrest, this compound is a potent inducer of apoptosis, or programmed cell death, in malignant cells drugbank.comnih.gov. This crucial mechanism contributes significantly to its therapeutic efficacy droracle.ainih.gov. This compound induces apoptosis in cells harboring the BCR-ABL fusion protein, such as those found in chronic myeloid leukemia (CML), and in gastrointestinal stromal tumor (GIST) cells that express an activating c-Kit mutation drugbank.comdroracle.ainih.gov. The induction of apoptosis is a direct consequence of this compound blocking the proliferative signals originating from these constitutively active tyrosine kinases nih.gov. Studies have demonstrated that this compound induces mitochondria-mediated apoptosis in gastric cancer cells, a process that involves endoplasmic reticulum (ER) stress-associated activation of c-Jun NH2-terminal kinase (JNK) spandidos-publications.com. Furthermore, this compound has been shown to induce cell death in osteosarcoma cell lines, with the mechanism being partly dependent on caspase activation plos.org. In breast cancer cell lines, significant apoptosis induction was observed after 96 hours of this compound treatment, following an initial period of cell cycle arrest dovepress.com. This indicates a time-dependent progression from cytostasis to cell death in some contexts dovepress.com.
Preclinical Research and Drug Discovery Paradigms of Imatinib
In Vitro Cellular Efficacy Studies
Selective Suppression of Oncogene-Expressing Cell Lines (e.g., BCR-ABL, K562)
Imatinib potently inhibits the growth of cells expressing the p210 BCR-ABL fusion protein, demonstrating selective antiproliferative activity ashpublications.org. This selective effect was confirmed across numerous cell lines derived from CML or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL), while Ph-negative cell lines remained unaffected ashpublications.org.
For instance, this compound exhibited significant growth inhibition in the K562 cell line, a widely used CML cell model that constitutively expresses BCR-ABL ashpublications.orgashpublications.orgjcancer.org. The inhibitory concentration 50 (IC50) for this compound in inhibiting proliferation generally mirrored its IC50 values for inhibiting BCR-ABL kinase activity in cellular assays ashpublications.org. Exposure to this compound ultimately leads to apoptotic cell death in these sensitive cell lines ashpublications.org.
It was observed that this compound potently inhibited all ABL tyrosine kinases, including cellular ABL, viral ABL (v-ABL), and BCR-ABL ashpublications.org. Studies using purified enzymes or immunoprecipitations showed that this compound inhibited ABL kinase activity with IC50 values ranging between 0.1 and 0.35 µM ashpublications.org.
| Cell Line Type | BCR-ABL Expression | This compound Effect | Outcome | Citation |
| CML/Ph+ ALL | Positive | Growth Inhibition | Apoptotic cell death | ashpublications.orgspandidos-publications.com |
| K562 | Positive | Potent Inhibition | Apoptotic cell death, reduced proliferation | ashpublications.orgashpublications.orgjcancer.orgspandidos-publications.com |
| Ph-negative | Negative | Unaffected | No growth inhibition | ashpublications.org |
Colony-Forming Unit Assays
Colony-forming unit (CFU) assays are crucial for assessing the impact of compounds on hematopoietic progenitor cells. This compound has been shown to inhibit the growth of colony-forming units, specifically granulocyte/macrophage colony-forming units (CFU-GM) and erythroid burst-forming units (BFU-E), in a dose-dependent manner ashpublications.org. This inhibition was observed even at low concentrations of this compound, such as 1.25 µM, which reduced the growth of CFU-GM in clonogenic assays ashpublications.orgnih.gov.
A reduction in colony-forming capacity due to this compound treatment was detectable from concentrations as low as 1.25 µM for total colonies, significantly affecting BFU-Es from 2.5 µM and CFU-GMs from 0.625 µM ashpublications.org. This compound treatment also led to a reduction in colony size compared to controls ashpublications.org. This effect on normal progenitor cells indicates that this compound can influence hematopoietic cell proliferation, although its primary clinical efficacy is directed at malignant BCR-ABL-positive cells bjournal.org.
| Cell Type | Assay Type | This compound Concentration (µM) | Effect on Colony Formation | Citation |
| CD34+ cells (normal) | Methylcellulose progenitor assay (CFU) | 1.25 | Detectable reduction in total colonies | ashpublications.org |
| CD34+ cells (normal) | Methylcellulose progenitor assay (CFU) | 0.625 | Significant reduction in CFU-GMs | ashpublications.org |
| CD34+ cells (normal) | Methylcellulose progenitor assay (CFU) | 2.5 | Significant reduction in BFU-Es | ashpublications.org |
In Vivo Efficacy Studies in Animal Models
Preclinical in vivo studies in animal models were instrumental in demonstrating this compound's potent antitumor activity against BCR-ABL-expressing cells at tolerated doses ashpublications.orgaacrjournals.org. These studies confirmed the selective action of this compound observed in vitro and provided crucial evidence for its potential as a therapeutic agent for CML.
For instance, this compound caused dose-dependent inhibition of tumor growth in syngeneic mice injected with BCR-ABL-transformed 32D cells ashpublications.org. Treatment with this compound (ranging from 2.5 to 50 mg/kg once daily intraperitoneal) starting one week after cell injection, significantly suppressed tumor growth ashpublications.org. Importantly, 50 mg/kg intraperitoneal treatment was found to be inactive against tumors derived from v-SRC-transformed 32D cells, which is consistent with this compound's lack of inhibition of SRC kinase activity ashpublications.org.
In a transduction-transplantation model of CML, where lethally irradiated syngeneic mice received bone marrow infected with a BCR-ABL retrovirus and consistently died within three weeks from aggressive CML, treatment with this compound (50 mg/kg in the morning, 100 mg/kg in the evening) led to prolonged survival ashpublications.org. Eight out of twelve animals remained tumor-free for over 200 days, while four relapsed between days 48 and 60 ashpublications.org. The antitumor effect of this compound was specific for p210BCR-ABL expressing cells, as no growth inhibition occurred in mice injected with U937, a BCR-ABL-negative myeloid cell line ashpublications.org.
Studies using human CML cell line K562 xenografts in nude mice also demonstrated dose-dependent inhibition of tumor growth and even tumor regressions with this compound treatment ashpublications.org. This included activity against human BCR-ABL-positive cell lines such as K562, highlighting its translational potential ashpublications.org. Animal models further showed that this compound effectively reduced the activation of BCR-ABL and downstream signaling in specimens derived from patients with this compound-sensitive CML tandfonline.com.
| Animal Model / Cell Line | BCR-ABL Status | This compound Treatment | Observed Outcome | Citation |
| Syngeneic mice + BCR-ABL-transformed 32D cells | Positive | 2.5-50 mg/kg IP, once daily | Dose-dependent inhibition of tumor growth | ashpublications.org |
| Syngeneic mice + v-SRC-transformed 32D cells | Negative | 50 mg/kg IP | Inactive against tumors | ashpublications.org |
| Syngeneic mice + BCR-ABL retrovirus-infected marrow | Positive | 50-100 mg/kg daily | Prolonged survival, tumor-free survival | ashpublications.org |
| Nude mice + K562 human CML xenografts | Positive | Dose-dependent | Dose-dependent tumor growth inhibition, tumor regression | ashpublications.org |
Structural Optimization and Analog Development
Design Principles for Novel this compound Analogues
The development of this compound was a prime example of rational drug design, initiated after the identification of the Philadelphia chromosome mutation and the hyperactive BCR-ABL protein wikipedia.orguniversiteitleiden.nl. The initial lead compound, a 2-phenylaminopyrimidine derivative, was identified through high-throughput screening wikipedia.orgpagepress.org. This compound, however, had low potency and poor specificity, inhibiting both serine/threonine and tyrosine kinases ashpublications.orgpagepress.org.
Subsequent structural optimization involved systematic modifications to enhance its selectivity and potency:
Enhancement of Cellular Activity: The addition of a 3′-pyridyl group at the 3′-position of the pyrimidine ring significantly enhanced the cellular activity of the derivatives ashpublications.orgpagepress.org.
Improved Tyrosine Kinase Activity: Introducing a benzamide group to the phenyl ring further enhanced activity against tyrosine kinases ashpublications.orgpagepress.org.
Reduced PKC Inhibition: A key observation from structure-activity relationship (SAR) analysis was that substitutions at the 6-position of the anilino phenyl ring led to a loss of protein kinase C (PKC) inhibition ashpublications.orgpagepress.org. The introduction of a "flag-methyl" group at this position either retained or enhanced activity against ABL tyrosine kinases ashpublications.orgpagepress.org.
Increased Solubility and Bioavailability: The attachment of an N-methylpiperazine group markedly improved water solubility and oral bioavailability, making it a clinically viable drug ashpublications.orgpagepress.org.
These design principles, leveraging medicinal chemistry and understanding of protein kinase binding sites, have guided the development of subsequent generations of ABL kinase inhibitors and novel this compound analogues nih.govnih.govaacrjournals.org. The goal is often to expand the structural diversity of BCR-ABL inhibitors and to overcome acquired resistance mechanisms, such as those arising from mutations in the BCR-ABL kinase domain benthamdirect.compnas.orgmdpi.com.
Structure-Activity Relationship (SAR) Studies of this compound Derivatives
SAR studies are crucial for understanding how modifications to a compound's chemical structure influence its biological activity, and these have been extensively performed for this compound and its derivatives benthamdirect.commdpi.comacs.org. These studies aim to identify key pharmacophoric features and optimize drug properties like potency, selectivity, and pharmacokinetic profiles.
For this compound, the 2-phenylaminopyrimidine scaffold is a fundamental pharmacophoric fragment mdpi.comresearchgate.net. SAR studies have explored modifications to various parts of the this compound molecule:
Benzene (A ring) and Piperidine (E ring) Substitutions: Introducing benzene and piperidine rings instead of the pyridine (A ring) and piperazine (E ring) in this compound has been shown to significantly enhance potency against K562 and HL-60 cell lines benthamdirect.com. This is consistent with docking results, suggesting improved interactions with the target benthamdirect.com.
Phenylamino Moiety Modifications: Substitutions in the -3 and -4 positions of the phenylamino moiety have led to improved potency and selectivity in both target-based and cell-based assays for certain pyrido[2,3-d]pyrimidin-7-one compounds, a class of Abl kinase inhibitors nih.gov.
Specific Group Importance: The importance of specific groups, such as the CF₂ group, has been highlighted in studies of new this compound derivatives, where its absence led to a loss of activity mdpi.com.
Overcoming Resistance: SAR studies have been instrumental in designing novel derivatives capable of overcoming this compound resistance, particularly mutations like T315I pnas.orgmdpi.com. For example, thiazolo[5,4-b]pyridine derivatives were synthesized, and SAR studies identified compounds with higher enzymatic and cellular activities against c-KIT mutants, including those resistant to this compound mdpi.com. One such derivative, 6r, demonstrated 8.0-fold higher enzymatic inhibitory activity against a c-KIT V560G/D816V double mutant and 23.6-fold higher antiproliferative activity on HMC1.2 cells than this compound mdpi.com.
The ongoing SAR research continues to provide useful information for the rational design of new compounds with improved properties, aiming to enhance efficacy and overcome resistance mechanisms in CML and other cancers where ABL or related kinases are implicated benthamdirect.commdpi.comijpsr.com.
Cytostatic Effects of this compound Analogues on Cancer Cell Lines
The development of chemical analogues of existing therapeutics, such as this compound, represents a crucial strategy in drug discovery. This approach aims to enhance potency, improve selectivity, overcome drug resistance, or reduce off-target effects. Preclinical research into this compound analogues has demonstrated diverse cytostatic effects across various cancer cell lines, offering insights into structure-activity relationships and potential future therapeutic candidates.
Research Findings
Several studies have explored the antiproliferative capabilities of novel this compound derivatives. One investigation synthesized new analogues incorporating isatins and the phenylamino-pyrimidine pyridine (PAPP) scaffold, which is a key pharmacophore of this compound nih.gov. These compounds were evaluated for their antiproliferative activity against human lung adenocarcinoma (A549) and chronic myeloid leukemia (K562) cell lines. Notably, certain 3,3-difluorinated this compound derivatives demonstrated significantly superior potency against A549 cells compared to this compound itself. Specifically, compounds 3a, 3c (5-chloro-3,3-difluorinated), and 3d (5-bromo-3,3-difluorinated) exhibited IC50 values of 7.2 µM, 6.4 µM, and 7.3 µM, respectively, while this compound's IC50 against A549 was 65.4 µM nih.gov. In contrast, for the K562 cell line, while this compound showed an exceptionally low IC50 of 0.08 µM, the derivative 3b (3,3-difluoro-5-methylated) had an IC50 of 35.8 µM at 10 µM, indicating that the potency against this cell line was not universally improved for all analogues nih.gov.
Another series of this compound derivatives, N-(2,5-dimethylphenyl)-4-pyridin-3-ylpyrimidin-2-amine compounds bearing a 2-chloroquinoline motif, were synthesized and screened for antiproliferative activity against human alveolar adenocarcinoma (A549) and human breast adenocarcinoma (MCF7) cell lines ijper.org. At a concentration of 10 µM, these compounds showed moderate cell growth inhibition. Among them, compound 4c, which features a dimethoxy group at the 6 and 8 positions of the 2-chloroquinoline ring, exhibited the highest antiproliferative activity, resulting in 35.21% cell viability for A549 and 30.36% for MCF7 cell lines ijper.org. This suggests that specific structural modifications can retain, and in some contexts, optimize the anticancer activity of this compound derivatives ijper.org.
Further research into this compound derivatives evaluated two compounds, 2a and 2b, against breast cancer (MCF-7) and colorectal cancer (HCT116) cell lines impactfactor.orgresearchgate.net. Compound 2b demonstrated superior activity against the HCT116 cell line with an IC50 of 15.88 µg/mL, compared to this compound's 18.52 µg/mL. However, against the MCF-7 cell line, both compounds 2a and 2b showed lower potency (IC50s of 127.35 µg/mL and 125.88 µg/mL, respectively) compared to this compound's 19.34 µg/mL impactfactor.orgresearchgate.net. Interestingly, compound 2b also exhibited improved cytotoxic activity on normal non-cancerous MDCK kidney cells, meaning it was less toxic to these cells compared to this compound, suggesting a potentially better therapeutic index impactfactor.orgresearchgate.net.
In the context of Bcr-Abl inhibition, new this compound analogues have been developed and assessed for their cytostatic effects on various human cancer cell lines, including K562 (chronic myeloid leukemia) and HL-60 (acute myeloid leukemia) benthamdirect.comresearchgate.net. One such analogue, compound 1h, displayed lower IC50 values for both K562 and HL-60 cells compared to this compound, indicating enhanced potency. Studies further revealed that compound 1h was superior to this compound in inhibiting Bcr-Abl expression and inducing pro-apoptotic effects in K562 cells benthamdirect.comresearchgate.net. Structural analysis indicated that replacing the pyridine (A ring) and piperazine (E ring) moieties in this compound with benzene and piperidine rings, respectively, significantly enhanced the potency of the analogue against K562 and HL-60 cell lines benthamdirect.comresearchgate.net.
Data Tables
The following tables summarize the cytostatic effects of various this compound analogues on different cancer cell lines, based on published research findings.
Table 1: Cytostatic Activity of this compound Derivatives (Isatin/PAPP Scaffold) on Cancer Cell Lines
| Compound | Cell Line | Effect (IC50) | This compound IC50 (for comparison) | Source |
| This compound Derivative 3a | A549 | 7.2 µM | 65.4 µM | nih.gov |
| This compound Derivative 3c | A549 | 6.4 µM | 65.4 µM | nih.gov |
| This compound Derivative 3d | A549 | 7.3 µM | 65.4 µM | nih.gov |
| This compound Derivative 3b | K562 | 35.8 µM | 0.08 µM | nih.gov |
Table 2: Cytostatic Activity of this compound Derivatives (Quinoline Scaffold) on Cancer Cell Lines
| Compound | Cell Line | Effect (% Cell Viability at 10 µM) | This compound (% Cell Viability at 10 µM) | Source |
| This compound Derivative 4c | A549 | 35.21 ± 3.67 % | Not explicitly reported in source | ijper.org |
| This compound Derivative 4c | MCF7 | 30.36 ± 2.40 % | Not explicitly reported in source | ijper.org |
Table 3: Cytostatic Activity of Other this compound Derivatives on Cancer Cell Lines
| Compound | Cell Line | Effect (IC50) | This compound IC50 (for comparison) | Source |
| This compound Derivative 2b | HCT116 | 15.88 µg/mL | 18.52 µg/mL | impactfactor.orgresearchgate.net |
| This compound Derivative 2a | MCF-7 | 127.35 µg/mL | 19.34 µg/mL | impactfactor.orgresearchgate.net |
| This compound Derivative 2b | MCF-7 | 125.88 µg/mL | 19.34 µg/mL | impactfactor.orgresearchgate.net |
| This compound Analogue 1h | K562 | Lower than this compound | Reported as "Lower than this compound" | benthamdirect.comresearchgate.net |
| This compound Analogue 1h | HL-60 | Lower than this compound | Reported as "Lower than this compound" | benthamdirect.comresearchgate.net |
Mechanisms of Imatinib Resistance in Research
BCR-ABL Dependent Resistance Mechanisms
BCR-ABL dependent mechanisms of imatinib resistance primarily involve qualitative or quantitative modifications of the BCR-ABL target protein researchgate.netresearchgate.net. These include point mutations within the ABL kinase domain and overexpression or amplification of the BCR-ABL gene researchgate.netresearchgate.netresearchgate.netaacrjournals.org.
ABL Kinase Domain Point Mutations
Point mutations within the ABL kinase domain of the BCR-ABL fusion protein are the most frequently identified mechanism of acquired resistance to this compound, observed in 30-50% of cases thieme-connect.comashpublications.orgnih.gov. Over 100 distinct amino acid substitutions have been identified, with around 40 different amino acid substitutions reported to date aacrjournals.orgbiorxiv.orgpnas.orgnih.gov. These mutations prevent this compound from binding effectively to its target without necessarily affecting ATP binding or kinase activity aacrjournals.orgehaweb.org. The frequency of these mutations varies with disease phase, ranging from 25-30% in early chronic phase (CP) CML patients on first-line this compound to approximately 70-80% in blast crisis (BC) patients ashpublications.org.
A table summarizing the common ABL kinase domain mutations and their impact on this compound resistance is provided below.
| Mutation Code | Location/Type | Frequency (%) (Approx.) aacrjournals.org | Resistance Level (General) | Notes |
| M244V | P-loop | 10 | Variable | One of the seven residues accounting for ~85% of resistance mutations aacrjournals.org. |
| G250E | P-loop | 10 | Markedly resistant nih.gov | Highly recurrent in advanced-stage CML and Ph+ ALL patients aacrjournals.org. One of the seven residues accounting for ~85% of resistance mutations aacrjournals.org. |
| Y253F/H | P-loop | 13 | Markedly resistant (Y253F) nih.gov, Partial (Y253H) | Highly recurrent in advanced-stage CML and Ph+ ALL patients aacrjournals.org. One of the seven residues accounting for ~85% of resistance mutations aacrjournals.org. Y253 is critical for efficient this compound binding ashpublications.org. |
| E255K/V | P-loop | 17 | Markedly resistant (E255K) nih.gov | Highly recurrent in advanced-stage CML and Ph+ ALL patients aacrjournals.org. One of the seven residues accounting for ~85% of resistance mutations aacrjournals.org. |
| T315I | This compound Binding Site | 12 | Complete resistance synnovis.co.uk | Known as the "gatekeeper" residue, crucial for this compound binding aacrjournals.orgehaweb.org. Confers resistance to all currently approved ABL kinase inhibitors pnas.org. Frequently found in advanced-stage CML and Ph+ ALL and associated with progression to accelerated phase/blast crisis aacrjournals.org. |
| M351T | Catalytic/Activation Loop | 11 | Moderate resistance nih.gov | One of the seven residues accounting for ~85% of resistance mutations aacrjournals.org. |
| F359V/I | Catalytic/Activation Loop | 11 | Variable | One of the seven residues accounting for ~85% of resistance mutations aacrjournals.org. F359V is more sensitive to dasatinib nih.gov. |
| H396P | Activation Loop | - | Variable | May stabilize the active conformation of the kinase jnccn.org. |
| Q252H | P-loop | - | Moderate resistance nih.gov | |
| F317L | Contact Site | - | Moderate resistance nih.gov | More sensitive to nilotinib nih.gov. |
| E355G | - | - | Moderate resistance nih.gov | |
| V280G | - | - | Potential resistance nih.gov | A newly described mutation, V280G, has been reported in an this compound-resistant CML patient nih.gov. |
Mutations Affecting this compound Binding Site
This compound functions by competitively targeting the ATP binding site within the ABL kinase domain, thereby stabilizing the kinase in a catalytically inactive conformation nih.govjnccn.orgpnas.org. Mutations affecting the this compound binding site can directly impair drug binding through steric hindrance or by destabilizing the inactive kinase conformation necessary for this compound binding aacrjournals.orgpnas.orgehaweb.orgpnas.org. The side chain of threonine 315 (T315) forms a hydrogen bond with this compound, making the T315I mutation particularly significant as it introduces an isoleucine, a bulkier amino acid, which sterically hinders this compound binding, leading to complete resistance aacrjournals.orgehaweb.orgjnccn.org. Other mutations, such as Y253F/H and E255K/V, also located in the ATP-binding site, can disrupt optimal drug interaction aacrjournals.orgashpublications.org.
P-loop (ATP Binding Site) Mutations
The P-loop (amino acids 248-256) is a highly conserved region of the ABL kinase domain responsible for ATP phosphate binding aacrjournals.orgjnccn.org. Mutations within the P-loop are the most frequently occurring this compound-resistant mutations, accounting for 36% to 48% of all mutations nih.gov. These mutations, such as G250E, Y253F/H, E255K/V, and M244V, often disrupt the proper positioning of the P-loop, preventing the kinase from adopting the inactive conformation required for this compound binding aacrjournals.orgnih.govjnccn.org. P-loop mutations are frequently observed in advanced-stage CML and Ph+ ALL patients and are often associated with progression from chronic phase to accelerated phase or blast crisis aacrjournals.org. Some P-loop mutations, like Y253F and E255K, have shown increased oncogenic potency relative to unmutated BCR-ABL and other mutated variants in in vitro studies nih.gov.
Catalytic (C) Domain and Activation Loop Mutations
Mutations can also occur in the catalytic (C) domain and the activation loop (A-loop) of the ABL kinase domain, impacting this compound binding and kinase activity nih.govsynnovis.co.uk. The A-loop is crucial for regulating kinase activity, and mutations like H396P can stabilize the active conformation of the kinase, which this compound is unable to bind jnccn.orgresearchgate.net. The M351T mutation, located at the base of the activation loop, is another common mutation that can confer resistance aacrjournals.orgjnccn.org. Mutations in these regions can alter the conformational dynamics of the protein, potentially leading to faster dissociation of this compound even if initial binding affinity is wild-type-like biorxiv.orgpnas.org.
Pre-existing Mutation Clones and Disease Phenotype
Research suggests that mutated BCR-ABL-expressing cells may pre-exist at low, undetectable levels in patients prior to this compound treatment nih.govunibo.itplos.org. These pre-existing clones can then expand under the selective pressure of this compound therapy, leading to acquired resistance and disease progression nih.govunibo.itresearchgate.net. The presence of such mutations, particularly those affecting drug binding directly or indirectly, has been associated with this compound resistance and disease progression researchgate.netamerihealth.com. Studies have used highly sensitive techniques like allele-specific oligonucleotide polymerase chain reaction (ASO-PCR) to detect rare mutated cells in pre-treatment samples, supporting the theory of pre-existing mutations nih.govresearchgate.net. The detection of pre-existing BCR-ABL mutations in CD34+ stem/progenitor cells of newly diagnosed chronic-phase CML patients has been linked to subsequent this compound resistance researchgate.net. While the T315I mutation is not necessarily more likely to pre-exist than other resistance mutations, T315I cells tend to establish larger clones at diagnosis plos.org. The "fitness" of these mutant clones, their ability to proliferate and gain a selective advantage, is highly relevant to the risk of resistance and disease progression unibo.itplos.org.
BCR-ABL Gene Amplification and Protein Overexpression
Beyond point mutations, BCR-ABL gene amplification and subsequent protein overexpression represent another significant BCR-ABL dependent mechanism of this compound resistance researchgate.netaacrjournals.orgresearchgate.netresearchgate.netaacrjournals.orgconicet.gov.ar. This mechanism can lead to elevated ABL kinase activity, potentially overwhelming the inhibitory capacity of this compound researchgate.netresearchgate.net. BCR-ABL gene amplification is more commonly observed in advanced-phase disease aacrjournals.org. Studies have reported that overexpression of BCR-ABL transcript can be associated with this compound resistance, with increased expression levels hindering the therapeutic efficacy of this compound researchgate.net. This overexpression can herald disease progression, where genetic instability is higher, and oncogenic addiction might be reduced researchgate.net. The extent of BCR-ABL transcript overexpression linked to this compound resistance has been estimated to range from a few-fold to one log or more researchgate.net.
Pharmacokinetic and Pharmacodynamic Research of Imatinib
Population Pharmacokinetics
The pharmacokinetics of imatinib can exhibit considerable inter-patient variability, necessitating a detailed understanding of its absorption, distribution, metabolism, and excretion within patient populations.
This compound is nearly completely absorbed, with oral bioavailability exceeding 97% aacrjournals.orgamegroups.cn. Peak plasma concentrations (Cmax) are typically achieved around 1.8 to 4 hours after a single dose and 1 to 6.8 hours at steady state iiarjournals.org. Upon chronic administration, systemic exposure to this compound tends to increase. For instance, in patients receiving a daily dose of 400 mg, the area under the concentration-time curve (AUC) over 24 hours (AUC0-24h) increased from 24.8 mg·h/L (SD: 7.4) on day 1 to 40.1 mg·h/L (SD: 15.7) at steady state iiarjournals.org. At steady state, with daily doses of 400 mg or 600 mg, Cmax typically ranges from 2.6 to 3.5 mg/L, and the terminal half-life is approximately 15 to 19 hours iiarjournals.org. The total body clearance is around 200 mL/min iiarjournals.org.
Population pharmacokinetic analyses have indicated a mean oral clearance of 14.3 L/h (± 1.0 SEM) and a volume of distribution of 347 L (± 62 SEM) nih.govnih.gov. Studies have also observed a decrease in this compound clearance from day 1 to day 29 by approximately 4 L/h, while the volume of distribution remained unchanged nih.gov. After more than one year of treatment, this compound clearance has been observed to increase by approximately 33%, leading to a decrease in systemic exposure by about 42% compared to the start of treatment aacrjournals.orgeur.nl.
Data on this compound's systemic exposure and clearance dynamics are summarized in Table 1.
| Pharmacokinetic Parameter (400 mg daily) | Day 1 | Steady State |
| AUC0-24h (mg·h/L) | 24.8 (SD: 7.4) iiarjournals.org | 40.1 (SD: 15.7) iiarjournals.org |
| Cmax (mg/L) | 1.9 (SD: 0.35) iiarjournals.org | 2.59 (SD: 0.78) iiarjournals.org |
| Tmax (h) | 1.8 - 4 iiarjournals.org | 1 - 6.8 iiarjournals.org |
| Terminal Half-life (h) | Not specified | 15 - 19 iiarjournals.org |
| Total Body Clearance (mL/min) | Not specified | ~200 iiarjournals.org |
| Oral Clearance (L/h) | 14.3 (± 1.0 SEM) nih.govnih.gov | Decreased by 4 (3-5) L/h nih.gov |
| Volume of Distribution (L) | 347 (± 62 SEM) nih.govnih.gov | Unchanged nih.gov |
Several factors contribute to the inter-individual variability in this compound pharmacokinetics. This compound is highly bound to plasma proteins, approximately 95%, predominantly to alpha-1 acid glycoprotein (AAG) aacrjournals.orgamegroups.cniiarjournals.org. AAG levels can vary substantially among patients, with a reported five-fold variation in CML patients, influencing the unbound fraction of this compound available for clearance and distribution to target cells amegroups.cn. Higher concentrations of AAG have been correlated with increased total this compound peak plasma concentrations at steady state iiarjournals.org. In animal models, elevated AAG levels were associated with an absence of response to this compound, suggesting that high AAG concentrations might decrease the active unbound fraction of the drug, thus limiting its access to target sites iiarjournals.org. While AAG levels significantly influence total this compound concentrations, the relationship between AAG levels and the AUC has not been consistently significant iiarjournals.orgaacrjournals.org.
Other demographic covariates, such as body weight, age, sex, and disease diagnosis, also influence oral clearance nih.govnih.gov. For instance, doubling body weight can be associated with a 12% increase in clearance nih.gov. However, a significant proportion of inter-individual variability (36% for clearance and 63% for volume of distribution) remains unexplained by these demographic factors nih.govnih.gov. This compound is primarily metabolized by cytochrome P450 (CYP) 3A4 and CYP3A5 enzymes, with minor contributions from CYP1A2, CYP2D6, CYP2C9, and CYP2C19 aacrjournals.orgfda.govaacrjournals.orguniversiteitleiden.nl. This reliance on CYP3A4 for metabolism indicates a potential for drug-drug interactions with CYP3A4 modulators fda.govaacrjournals.org.
| Factor | Influence on this compound Pharmacokinetics | References |
| Alpha-1 Acid Glycoprotein (AAG) | High plasma AAG concentrations correlate positively with total this compound peak plasma concentrations at steady state. AAG levels contribute to inter-individual variability in total this compound clearance and can affect the unbound fraction of the drug amegroups.cniiarjournals.orgnih.govnih.govaacrjournals.org. | amegroups.cniiarjournals.orgnih.govnih.govaacrjournals.org |
| Body Weight | Influences oral clearance and volume of distribution; doubling body weight can increase clearance by 12% nih.govnih.govnih.gov. | nih.govnih.govnih.gov |
| Age | Influences oral clearance nih.govnih.gov. | nih.govnih.gov |
| Sex | Influences oral clearance nih.govnih.gov. | nih.govnih.gov |
| Disease Diagnosis | Influences oral clearance nih.govnih.gov. | nih.govnih.gov |
| CYP3A4/3A5 Activity | Major enzymes responsible for this compound metabolism; modulators (inducers/inhibitors) of these enzymes can affect this compound exposure aacrjournals.orgfda.govaacrjournals.orguniversiteitleiden.nl. | aacrjournals.orgfda.govaacrjournals.orguniversiteitleiden.nl |
| Long-term Treatment (>1 year) | Can lead to an increase in this compound clearance (~33%) and a decrease in systemic exposure (~42%) aacrjournals.orgeur.nl. | aacrjournals.orgeur.nl |
| Volume of Liver Metastases (in GIST) | A predicted decrease of 3.8% in this compound clearance for every 100 cm³ increase of metastatic volume was observed in GIST patients, though this effect is considered marginal nih.gov. | nih.gov |
| Creatine Kinase (CK) Levels | Patients with higher CK levels exhibit higher this compound peak and trough plasma concentrations. For example, high CK group showed mean Cmax of 3166.5 ng/mL vs. 2665 ng/mL (low CK group), and trough Cmin of 1422 ng/mL vs. 1169.5 ng/mL (low CK group) nih.gov. | nih.gov |
Systemic Exposure and Clearance Dynamics
Metabolite Pharmacokinetics and Activity
This compound is metabolized primarily to N-desmethyl this compound, also known as CGP74588. This metabolite is predominantly formed via CYP3A4, with minor contributions from CYP3A5 and CYP2D6 aacrjournals.orgiiarjournals.orgfda.govaacrjournals.orguniversiteitleiden.nl. CGP74588 is the main circulating metabolite of this compound aacrjournals.orguniversiteitleiden.nl. The systemic exposure to CGP74588 typically represents approximately 10% to 15% of that for this compound aacrjournals.orguniversiteitleiden.nl. At steady state, the mean trough concentration (Cmin) for CGP74588 has been reported as 242 ng/mL (± 106 ng/mL, 43.6% CV) ashpublications.orgcapes.gov.br.
Importantly, CGP74588 exhibits in vitro activity comparable to that of the parent drug, this compound, in inhibiting tyrosine kinases aacrjournals.orgiiarjournals.orgfda.gov. While CGP74588 is an active metabolite, some studies have suggested it may be significantly less active in specific contexts nih.gov. In vitro studies indicate that CGP74588 can inhibit its own formation and also inhibit CYP3A4/5, CYP2C9, and CYP2D6 fda.gov.
Pharmacokinetic-Pharmacodynamic Correlations
Understanding the correlation between this compound’s pharmacokinetic parameters and its pharmacodynamic effects is crucial for optimizing treatment outcomes.
Numerous studies have investigated the relationship between this compound plasma concentrations and clinical responses, particularly in chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST). In CML patients, higher steady-state this compound plasma levels have been significantly correlated with improved rates of both complete cytogenetic response (CCyR) and major molecular response (MMR) nih.govashpublications.orgcapes.gov.brnih.govd-nb.info.
Specifically, a subanalysis of the IRIS study involving 351 patients with chronic-phase CML found that this compound plasma trough concentrations (Cmin) greater than 1000 ng/mL were significantly associated with better cytogenetic and molecular responses ashpublications.orgcapes.gov.brnih.gov. Patients who achieved CCyR had a significantly higher mean this compound Cmin (1009 ± 544 ng/mL) compared to those who did not (812 ± 409 ng/mL; P = .01) ashpublications.orgcapes.gov.br. Similarly, cumulative estimated CCyR and MMR rates differed significantly among quartiles of this compound trough levels (P = .01 for CCyR, P = .02 for MMR) ashpublications.orgcapes.gov.br. Higher drug exposure correlates with increased frequencies of CCyR and MMR nih.gov. This suggests that maintaining adequate this compound plasma concentrations is important for achieving good clinical responses ashpublications.orgcapes.gov.br.
For GIST patients, studies have also shown an association between minimal (trough) plasma concentrations of this compound after one month of treatment and clinical benefit aacrjournals.orgeur.nlnih.gov. A lower response rate or shorter time to progression may occur when this compound plasma levels fall below approximately 1000 ng/mL in both CML and GIST aacrjournals.orgeur.nl.
Data on the relationship between this compound Cmin and response rates are provided in Table 3.
| Response Type | This compound Cmin (ng/mL) in Responders (Mean ± SD) | P-value (vs. Non-responders) | Reference |
| Complete Cytogenetic Response (CCyR) | 1009 ± 544 (achieved CCyR) | 0.01 | ashpublications.orgcapes.gov.br |
| 812 ± 409 (did not achieve CCyR) | |||
| Major Molecular Response (MMR) | Significant correlation with Cmin quartiles | 0.02 | ashpublications.orgcapes.gov.br |
Biomarkers for Imatinib Response Prediction and Prognosis
Genetic and Genomic Biomarkers
Genetic and genomic variations play a significant role in modulating a patient's response to imatinib, reflecting inherent biological differences that influence drug efficacy and disease progression.
Recent research has identified specific immune-related gene expression signatures as potential predictive biomarkers for this compound response in chronic myeloid leukemia (CML). A study aimed at identifying immune-related genes associated with this compound response in CML patients leveraged gene expression profiles from the Gene Expression Omnibus (GEO) database nih.govresearchgate.netdntb.gov.ua. Through bioinformatics analysis and weighted gene co-expression network analysis (WGCNA), 428 differentially expressed immune-related genes were identified, involved in pathways such as T-cell receptor signaling and cytokine-cytokine receptor interaction nih.govresearchgate.netdntb.gov.uaresearchgate.net.
Three hub genes, IL10RA , SCN9A , and SLC26A11 , emerged as potential biomarkers for predicting this compound response based on Receiver Operating Characteristic (ROC) analysis in two GEO datasets nih.govresearchgate.netdntb.gov.uaresearchgate.net. These findings were further validated in an independent clinical cohort of 60 CML patients treated with this compound nih.govresearchgate.net. Quantitative real-time polymerase chain reaction (qRT-PCR) results indicated that high expression of IL10RA and SLC26A11 was observed in responders, while SCN9A showed low expression nih.govresearchgate.netdntb.gov.ua. All three genes demonstrated an area under the curve (AUC) greater than 0.75, confirming their potential as predictive biomarkers nih.govresearchgate.netdntb.gov.ua.
Table 1: Immune-Related Gene Expression as Predictive Biomarkers for this compound Response in CML
| Gene Name | Expression in Responders | AUC | Pathway Involvement | Reference |
| IL10RA | High | >0.75 | T-cell receptor signaling, cytokine-cytokine receptor interaction | nih.govresearchgate.netdntb.gov.ua |
| SCN9A | Low | >0.75 | T-cell receptor signaling, cytokine-cytokine receptor interaction | nih.govresearchgate.netdntb.gov.ua |
| SLC26A11 | High | >0.75 | T-cell receptor signaling, cytokine-cytokine receptor interaction | nih.govresearchgate.netdntb.gov.ua |
MicroRNA (miRNA) dysregulation is a critical aspect of tumorigenesis and has been linked to cancer development, progression, and treatment response nih.gov. In the context of this compound therapy for CML, specific miRNA expression profiles have shown promise as biomarkers.
Research has correlated the expression levels of miR-21, miR-26b, and miR-451 with response to tyrosine kinase inhibitor (TKI) treatment nih.gov. Notably, at diagnosis, miR-451 levels were significantly higher in patients who achieved an optimal response after 6 and 12 months of therapy nih.govresearchgate.net. MiR-451 functions as a tumor suppressor and has been observed to be downregulated in various neoplasias, including CML, where it directly targets ABL and BCR-ABL1 nih.govresearchgate.net. This compound treatment can lead to the upregulation of miR-451 spandidos-publications.com.
Table 2: MicroRNA Expression Profiles and this compound Response in CML
| MicroRNA | Expression in Optimal Responders (at diagnosis) | Role/Function | Association with this compound Response | Reference |
| miR-21 | Lower | Oncogene | Higher levels in non-responders; decreased by this compound in CML nih.govresearchgate.netmdpi.com | nih.govresearchgate.netmdpi.com |
| miR-451 | Higher | Tumor Suppressor | Higher levels in responders; upregulated by this compound nih.govresearchgate.netspandidos-publications.com | nih.govresearchgate.netspandidos-publications.com |
| miR-26b | (Expressed, but less predictive power compared to miR-21 and miR-451) | (Not specified in predictive context) | Not consistently identified as a primary predictor of optimal response compared to miR-21 and miR-451 nih.govresearchgate.net | nih.govresearchgate.net |
Germline genetic variations contribute to the heterogeneity of patient responses to this compound in CML. Specifically, single nucleotide polymorphisms (SNPs) in genes such as ASXL1 and BIM have been identified as significant predictive biomarkers.
The ASXL1 rs4911231 and BIM rs686952 variants have been found to be independent predictors of various molecular responses (early molecular response, major molecular response, deep MRs including MR4 and MR4.5) and failure-free survival (FFS) in CML patients treated with this compound researchgate.netnih.gov. Research indicates that the homozygous T genotype for ASXL1 rs4911231 and the A allele for BIM rs686952 are associated with superior treatment outcomes researchgate.net.
MicroRNA Expression Profiles (e.g., miR-21, miR-26b, miR-451)
Cellular and Functional Biomarkers
Beyond genetic predispositions, cellular and functional characteristics of leukemic cells provide direct insights into their sensitivity to this compound.
The in vitro Colony Forming Cell (CFC) assay is a functional biomarker used to assess the proliferative capacity of hematopoietic stem and progenitor cells, including those from CML patients. This assay provides a direct measure of the sensitivity of leukemic cells to therapeutic agents like this compound.
This compound has been shown to selectively inhibit the colony formation of committed progenitor cells from CML patients, while demonstrating minimal effects on normal hematopoiesis at clinically relevant concentrations ashpublications.org. Studies have utilized CFC assay outputs as valuable predictive biomarkers researchgate.net. For instance, comparisons of this compound-treated CML CD34+ cells with untreated cells or those exposed to second-generation TKIs such as dasatinib have revealed reduced CFC output following this compound exposure oncotarget.comnih.gov.
Functional in vitro endpoints, including CFC assays, are considered important for identifying and characterizing CML stem cells and for predicting patient responses to specific therapeutic modalities, as well as anticipating disease relapse or progression capes.gov.br. The outputs from CFC assays have been analyzed alongside clinical information, such as White Blood Cell (WBC) counts and Sokal scores, in studies aiming to develop multivariable biomarker panels for predicting this compound response researchgate.net.
Immunomodulatory and Off-target Effects of Imatinib
Effects on Normal Hematopoiesis and Immune Cell Populations
Imatinib's impact extends to the normal hematopoietic system and various immune cell subsets, leading to diverse immunomodulatory effects.
Hematopoietic Progenitor Cell Mobilization, Proliferation, and Differentiation
This compound has been observed to affect the mobilization, proliferation, and differentiation of hematopoietic progenitor cells nih.govscielo.brnih.govoup.combjournal.org. Studies indicate a dose-dependent decrease in the proliferation potential of CD34+ cells when exposed to this compound in vitro ashpublications.org. This inhibition is also reflected in a reduced colony-forming capacity, affecting both erythroid (BFU-E) and granulocyte/macrophage (CFU-GM) progenitor cells ashpublications.org. This effect on progenitor cells is a contributing factor to the myelosuppression observed in some treated patients nih.govscielo.broup.com. The inhibition of c-Kit signaling, a target of this compound, may partly explain the reduction in CFU-GM colony growth, as Stem Cell Factor (SCF) acting via c-Kit synergizes with other growth factors to stimulate progenitor cell formation scielo.brbjournal.org.
Table 1: Effects of this compound on Hematopoietic Progenitor Cells (in vitro) ashpublications.org
| Cell Type/Assay | This compound Concentration (μM) | Effect |
| CD34+ cells | Dose-dependent | Decreased proliferation potential |
| CFU-GM colonies | 0.625, 1.25 | Dose-dependent decrease in colony-forming capacity |
| BFU-E colonies | 2.5 | Significant reduction in colony-forming capacity |
| Colony Size | Increasing doses | Reduction in colony size |
Impact on Hematopoietic Stem Cells
While this compound significantly affects hematopoietic progenitor cells, it generally leaves more primitive hematopoietic stem cells (HSCs) unaffected, or less affected, at standard therapeutic concentrations nih.govnih.govoup.comashpublications.org. However, some studies suggest that this compound might induce activation, expansion, and maturation of HSCs and multipotent hematopoietic progenitors (MPP1-4) in the bone marrow, especially at certain doses nih.govplos.org. Despite this, the most primitive, quiescent Philadelphia-positive (Ph+) leukemic stem cells (LSCs) often remain largely insensitive to this compound, contributing to disease persistence and potential relapse mdpi.comoncotarget.comashpublications.org.
Influence on Regulatory T Cells (Tregs)
This compound has a notable influence on regulatory T cells (Tregs), a critical population involved in maintaining peripheral tolerance. Clinical observations and preclinical studies indicate that this compound can inhibit the number and suppressive function of Tregs nih.govjst.go.jprupress.orgresearchgate.netspandidos-publications.com. This compound has been shown to impair Treg immunosuppressive function and FoxP3 expression, a key transcription factor for Treg lineage commitment, in vitro nih.gov. This effect is partly mediated by the drug's ability to reduce the activation of transcription factors STAT3 and STAT5 in Tregs, and to inhibit the phosphorylation of ZAP70 and LAT in the Treg T-cell receptor (TCR)-induced signaling cascade nih.gov. Furthermore, this compound's inhibition of lymphocyte-specific protein tyrosine kinase (LCK) appears to selectively induce apoptosis in effector Tregs (eTregs), which express lower levels of LCK and ZAP-70 compared to other T cells, making them more susceptible to signal-deprived apoptosis upon TCR signal inhibition rupress.orgbmj.com. This selective depletion of eTregs can enhance anti-tumor immune responses rupress.org.
Table 2: this compound's Effects on Regulatory T Cells (Tregs) nih.govrupress.org
| Target/Mechanism | Effect of this compound | Outcome |
| Treg immunosuppressive function | Impaired | Reduced peripheral tolerance, enhanced anti-tumor immunity |
| FoxP3 expression | Reduced | Affects Treg lineage commitment and function |
| STAT3 and STAT5 activation | Reduced phosphorylation | Attenuates FoxP3 expression |
| ZAP70 and LAT phosphorylation | Inhibited (in TCR signaling) | Interferes with Treg TCR-signaling pathway |
| LCK inhibition | Selective apoptosis of effector Tregs (eTregs) | Augments anti-tumor immunity by depleting suppressive cells |
Modulation of Antigen-Presenting Cells (APCs) Function and Differentiation
The effects of this compound on antigen-presenting cells (APCs) are complex and, at times, appear contradictory across different studies nih.gov. Some research suggests that this compound can negatively modulate dendritic cell (DC) development and down-regulate their antigen-presenting function nih.govashpublications.org. For example, in vitro exposure of human CD34+ progenitors to this compound has been shown to result in DCs with reduced expression of co-stimulatory molecules (CD80, CD40) and an inability to induce primary cytotoxic T lymphocyte (CTL) responses ashpublications.org. This compound may also inhibit the in vitro development of monocyte/macrophage lineages from normal human bone marrow progenitor cells ashpublications.org. Conversely, other studies report that this compound may enhance APC function and overcome tumor-induced CD4+ T-cell tolerance ashpublications.orgpnas.org. Treatment of APCs with this compound in vitro has been shown to enhance the activation of naive antigen-specific T cells and restore the responsiveness of tolerant T cells from tumor-bearing hosts ashpublications.org. This enhancement may be linked to the inhibition of c-kit phosphorylation in APCs ashpublications.org. This compound's influence on APCs can also involve modulating pathways like NF-κB signaling, which affects cytokine production, including IL-6 and TNF-α pnas.orgnih.gov.
Inhibition of Effector T Lymphocyte Functions
This compound has been documented to inhibit the proliferation and activation of both CD4+ and CD8+ effector T lymphocytes nih.govnih.govoup.comresearchgate.netresearchgate.netnih.govresearchgate.net. This inhibitory effect is dose-dependent and has been observed at therapeutically relevant concentrations researchgate.netresearchgate.netnih.gov. This compound can reduce antigen-specific interferon-gamma (IFN-γ) secretion from both CD4+ and CD8+ T-effector cells without necessarily causing T-cell viability loss researchgate.netnih.govresearchgate.net. The drug's influence on T-cell activation extends to reducing the expression of activation markers on CD3+ T cells researchgate.net. This modulation of T-cell function is attributed, in part, to this compound's interference with the T-cell receptor (TCR)-induced ZAP70 signaling pathway, potentially by targeting the tyrosine kinase Lck nih.govresearchgate.net. Notably, this inhibition of T-cell proliferation appears to be reversible upon removal of the drug researchgate.netnih.gov.
Table 3: Impact of this compound on Effector T Lymphocyte Functions researchgate.netnih.govresearchgate.netnih.gov
| T Cell Type | Function Affected | Specific Effect | Reversibility |
| CD4+ T cells | Proliferation & Activation | Inhibition in a dose-dependent manner | Reversible |
| IFN-γ secretion | Reduced antigen-specific IFN-γ secretion | Not directly affected | |
| CD8+ T cells | Proliferation & Activation | Inhibition in a dose-dependent manner | Reversible |
| IFN-γ secretion | Reduced antigen-specific IFN-γ secretion | Not directly affected | |
| Lytic Function | Inhibition of lysis of target cells | Reversible |
Cytotoxic T Cell Induction and Impairment
The induction of specific cytotoxic T cells (CTLs) appears to be impaired in chronic myeloid leukemia (CML) patients treated with this compound compared to those receiving interferon-alpha nih.govnih.govoup.comresearchgate.netcapes.gov.br. While primary CTL responses to viral infections like lymphocytic choriomeningitis virus (LCMV) may not be significantly impaired by this compound, the secondary expansion of antigen-experienced memory CTLs is reduced both in vitro and in vivo capes.gov.brnih.gov. This reduction in memory CTL expansion can lead to impaired protection against reinfection and a delayed onset of CTL-induced pathologies in disease models capes.gov.brnih.gov. This effect is particularly significant in the context of anti-leukemic immunity, where the long-term control of leukemia often relies on robust, persistent T-cell responses plos.org.
Natural Killer (NK) Cell Activity Modulation
This compound's influence on Natural Killer (NK) cells presents a nuanced picture. While some studies suggest that NK cells generally exhibit high resistance to the cytotoxic effects of this compound at clinically relevant concentrations, other research indicates modulatory effects on NK cell activity aai.org. For instance, this compound has been observed to increase the expression of NKG2D, an activating receptor on NK cells, and to decrease the production and release of MICA protein, a ligand for NKG2D frontiersin.org. This action contributes to the restoration of functional NKG2D signaling, which is crucial for NK cell cytotoxicity frontiersin.org.
However, conflicting findings exist, where this compound treatment, particularly in Bcr/Abl-expressing target cells, has been shown to decrease their susceptibility to NK-mediated lysis. This effect can involve the modulation of NKG2D ligands, alteration of GM1 expression, and interference with immunological synapse formation stemcell.com. Specifically, this compound may induce surface GM1 ganglioside on Bcr/Abl transfectants, which can prevent the proper redistribution of MHC-related antigen molecules in lipid rafts, thus hindering NK cell interaction stemcell.com. Additionally, this compound can affect cell surface glycosylation and decrease the expression of MHC-related antigens-A/B and UL-16-binding proteins on Bcr/Abl transfectants, thereby inhibiting the NKG2D activating pathway and NK-mediated cytolysis stemcell.com.
Despite these complexities, certain reports indicate that this compound can upregulate the expression of various activating receptors on NK cells, including NCR and NKG2D, and may enhance interferon-gamma (IFN-γ) release, thereby contributing to antitumor responses mdpi.com. This compound has also been shown to increase the surface expression of CXCR4, a chemokine receptor, on NK cells and monocytes in patients receiving the drug for CML, while reducing CXCR3 expression in NK cells aai.orgresearchgate.net. These modulations in chemokine receptor repertoire can influence immune cell trafficking.
Here's a summary of this compound's observed effects on NK cells:
| Effect Category | Specific Observation | Reference |
| Receptor Modulation | Increases NKG2D expression | frontiersin.orgmdpi.com |
| Increases CXCR4 expression | aai.orgresearchgate.net | |
| Decreases CXCR3 expression | aai.org | |
| Ligand Modulation | Decreases MICA protein production and release | frontiersin.org |
| Can decrease MHC-related Ags-A/B and UL-16-binding protein expression on Bcr/Abl targets | stemcell.com | |
| Functional Impact | Contributes to normal NK cytotoxicity through NKG2D signaling restoration | frontiersin.org |
| Can hamper NK/target immunological synapse formation (with Bcr/Abl targets) | stemcell.com | |
| May enhance IFN-γ release | mdpi.com | |
| NK cells generally resistant to cytotoxic effects | aai.org |
Molecular Mechanisms of Immunological Modulation
This compound exerts its immunomodulatory effects through various molecular mechanisms, including the inhibition of specific kinases and the modulation of inflammasome activation.
This compound, while primarily a BCR-ABL inhibitor, has "off-target" effects that include the inhibition of lymphocyte-specific protein tyrosine kinase (LCK) rupress.orgrupress.org. LCK is a crucial signaling molecule in T cells, playing a central role in T cell receptor (TCR) signaling jci.orgrupress.orgnih.gov. Effector regulatory T (eTreg) cells, which suppress immune responses, are intrinsically susceptible to LCK inhibition due to their comparatively lower expression of LCK and ZAP-70 (another key signaling molecule) rupress.orgnih.govresearchgate.net. The inhibition of LCK by this compound further reduces the TCR signal intensity in eTreg cells, rendering them selectively prone to apoptosis due to signal deprivation rupress.orgrupress.orgnih.govresearchgate.netbmj.com. This selective depletion of eTreg cells by this compound is a significant mechanism by which the drug can augment anti-tumor immunity rupress.orgresearchgate.net.
This compound has been shown to trigger the activation of the NOD-like receptor protein 3 (NLRP3) inflammasome and subsequent production of pro-inflammatory cytokines, particularly Interleukin-1β (IL-1β) biorxiv.orgresearchgate.net. The NLRP3 inflammasome is a multiprotein complex that plays a critical role in innate immune responses by processing and secreting inflammatory cytokines like IL-1β and IL-18 biorxiv.orgwikipedia.orgguidetopharmacology.orgmdpi.com.
Research indicates that this compound, at certain concentrations (e.g., 20 µM for 2-3 hours), can induce robust, NLRP3-dependent IL-1β production in myeloid cells such as human PMA-differentiated THP-1 cells and LPS-primed murine bone marrow-derived macrophages biorxiv.org. This activation is linked to lysosomal damage-associated cell lysis and potassium (K+) efflux, which are known triggers for NLRP3 inflammasome activation biorxiv.org. While this compound can induce a form of lytic cell death independently of the inflammasome, the maturation and secretion of IL-1β are strongly dependent on NLRP3 and caspase-1 biorxiv.org. Studies suggest that this compound-induced oxidative stress and the activation of nuclear factor kappa B (NF-κB) contribute to the activation of NLRP3 inflammasomes, leading to caspase-1 cleavage and IL-1β release researchgate.netresearchgate.net.
LCK Kinase Inhibition
Antitumor Immunity Augmentation
Beyond its direct effects on cancer cells, this compound contributes to augmenting antitumor immunity through several mechanisms, including enhancing effector T cell infiltration and influencing the dynamics of anti-leukemia T cell responses.
This compound can promote the infiltration of effector T cells, particularly CD8+ T cells, into the tumor microenvironment nih.govjst.go.jpresearchgate.net. This effect is observed in certain tumor types, such as CT26 colon cancer, where this compound treatment significantly elevated the expression of CD8 and CD8+ T cell-recruiting chemokine genes nih.govjst.go.jp. An increase in the frequency of interferon-γ+ (IFN-γ+) CD8+ T cells in this compound-treated tumors suggests an induction of antitumor immunity nih.govjst.go.jp.
The ability of this compound to selectively deplete effector regulatory T (eTreg) cells also contributes significantly to this augmentation rupress.orgresearchgate.netresearchgate.net. By inhibiting LCK, this compound induces apoptosis in eTreg cells, which are critical suppressors of immune responses against tumors rupress.orgrupress.orgnih.govresearchgate.netbmj.com. This depletion of immune-suppressive cells allows for the expansion of tumor antigen-specific CD8+ T cells, thereby enhancing T cell-mediated anti-tumor immune responses rupress.orgnih.govresearchgate.netresearchgate.net.
Mathematical models and experimental data suggest that this compound treatment in chronic myelogenous leukemia (CML) patients may promote the development of anti-leukemia immune responses as patients achieve remission nih.govnih.govresearchgate.netplos.org. These models propose a concept of an "optimal load zone" for leukemic cells, where the anti-leukemia immune response is most effective nih.govnih.govresearchgate.netplos.org.
However, this compound therapy can sometimes drive leukemic cell populations to fall below this optimal load zone too rapidly, which may not allow sufficient time to sustain the anti-leukemia T cell response nih.govnih.govplos.orgplos.org. This highlights a dynamic interplay between drug-induced leukemic cell reduction and the immune system's ability to mount a sustained response. The persistence of anti-leukemia T cells, even at low levels, appears to be crucial for preventing leukemia relapse and maintaining remission under this compound therapy nih.govnih.govresearchgate.net. Therefore, potential therapeutic strategies, such as combining this compound with vaccination approaches, are being explored to optimally sustain the anti-leukemia T cell response and potentially eradicate residual leukemic cells for a more durable cure nih.govnih.govplos.org.
Here's a table summarizing research findings related to Antitumor Immunity Augmentation:
| Mechanism | Research Finding | Reference |
| Effector T Cell Infiltration | Elevated expression of CD8 and CD8+ T cell-recruiting chemokine genes in CT26 colon cancer treated with this compound. | nih.govjst.go.jp |
| Increased frequency of IFN-γ+ CD8+ T cells in this compound-treated CT26 tumors. | nih.govjst.go.jp | |
| Selective depletion of effector T reg (eTreg) cells by this compound, leading to expansion of tumor antigen-specific CD8+ T cells. | rupress.orgnih.govresearchgate.netbmj.comresearchgate.net | |
| Optimal Load Zone Dynamics | This compound therapy can drive leukemic cell populations below an "optimal load zone" too quickly, potentially limiting sustained anti-leukemia T cell response. | nih.govnih.govplos.orgplos.org |
| Persistence of anti-leukemia T cells, even at low levels, is important for maintaining remission. | nih.govnih.govresearchgate.net | |
| Combination therapies (e.g., this compound + vaccination) may sustain anti-leukemia T cell response for durable cure. | nih.govnih.govplos.org |
Research on Imatinib in Diverse Disease Contexts Mechanistic Insights
Osteosarcoma Research
Imatinib's efficacy in osteosarcoma has been investigated through its interaction with key molecular targets and signaling pathways that drive tumor progression.
PDGFRα as a Molecular Target
Platelet-Derived Growth Factor Receptor alpha (PDGFRα) has been identified as a significant molecular target for this compound mesylate in osteosarcoma plos.orgnih.govnih.govresearchgate.net. Studies utilizing a phospho-receptor tyrosine kinase array kit have revealed PDGFRα as one of the main molecular targets for this compound mesylate, alongside other receptors such as PDGFRβ, Axl, RYK, EGFR, EphA2, EphA10, and IGF1R plos.orgnih.govresearchgate.netresearchgate.net. The functional activity of PDGFR in osteosarcoma cells (human MG-63, HOS; murine POS-1, MOS-J) has been confirmed through investigations of PDGF-BB induced signaling plos.orgnih.gov. This compound mesylate, at a concentration of 25 µM, markedly inhibited the PDGF-BB induced phosphorylation cascades of both PDGFRα and PDGFRβ, confirming their roles as functional targets in osteosarcoma plos.orgnih.gov. PDGFRα expression has been observed frequently (79.6%) in osteosarcoma patient surgical specimens and correlated with inferior event-free survival, highlighting its potential as a prognostic marker and therapeutic target nih.gov.
Table 1: PDGFRα and PDGFRβ Expression in Osteosarcoma Patients
| Receptor | Expression Frequency | Correlation with Inferior Event-Free Survival |
| PDGF-alpha | 79.6% | Yes (P < 0.05) |
| PDGF-beta | 86% | No (P = 0.15) |
| PDGF-AA | 80.4% | Yes (P < 0.05) |
| PDGF-B-B | 75.4% | No (P = 0.15) |
Inhibition of AKT/mTOR Signaling Pathway
This compound mesylate has demonstrated its ability to inhibit the AKT/mTOR signaling pathway in osteosarcoma cells plos.orgnih.gov. In both human (HOS) and mouse (MOS-J) osteosarcoma cell lines, this compound mesylate effectively inhibited the phosphorylation of mTOR and Akt, key components of this pathway plos.orgnih.gov. This inhibition contributes to the observed anti-proliferative effects of this compound in osteosarcoma cells, leading to a G0/G1 cell cycle arrest in most cell lines and inducing cell death nih.gov. The suppression of the PI3K/AKT/mTOR pathway by this compound has significant implications as its dysregulated activation contributes to cell survival and proliferation in various cancers researchgate.net.
Activation of ERK1/2 Phosphorylation
Beyond inhibiting pro-survival pathways, this compound mesylate also activates ERK1/2 phosphorylation in osteosarcoma cells plos.orgnih.gov. While this compound inhibits the PDGF-BB induced phosphorylation of PDGFRα and PDGFRβ, it also significantly reduces the PDGF-BB induced phosphorylation of Akt and ERK1/2, indicating a complex modulation of downstream signaling pathways plos.orgnih.govspandidos-publications.com. This dual effect, inhibiting certain pro-proliferative pathways while activating others, underscores the intricate mechanistic actions of this compound.
Systemic Sclerosis Research (Focus on Fibrosis Mechanisms)
This compound has been extensively investigated for its antifibrotic properties, particularly in the context of systemic sclerosis (SSc), a disease characterized by progressive fibrosis.
Antifibrotic Mechanisms
This compound acts as a potent antifibrotic agent by simultaneously targeting two major profibrotic pathways: the c-Abl kinase and the Platelet-Derived Growth Factor Receptor (PDGFR) smw.chuzh.choup.comnih.gov. The c-Abl tyrosine kinase is an important downstream signaling molecule of transforming growth factor beta (TGF-β), a key mediator of fibrosis oup.comnih.gov. This compound binds competitively to the ATP-binding pocket of c-Abl, effectively blocking its tyrosine kinase activity oup.com. In cells deficient for c-Abl, the induction of extracellular matrix (ECM) proteins by TGF-β is significantly decreased oup.com.
In experimental models, this compound has been shown to efficiently inhibit the development of bleomycin-induced dermal fibrosis and also reduce established fibrosis smw.chuzh.chresearchgate.net. It prevents the differentiation of resting fibroblasts into myofibroblasts and reduces the synthesis and accumulation of extracellular matrix in lesional skin smw.chuzh.ch. Studies have demonstrated that this compound, at pharmacologically relevant concentrations, inhibits the synthesis of collagen 1a1, collagen 1a2, and fibronectin-1 in SSc fibroblasts by up to 90% smw.choup.comresearchgate.net. Furthermore, this compound abrogates the stimulation of fibrotic gene expression, including fibronectin, connective tissue growth factor, plasminogen activator inhibitor-1, and tissue inhibitor of metalloproteinases, induced by TGF-β nih.gov. This broad inhibition of profibrotic mediators positions this compound as a promising compound for managing fibrotic diseases.
Table 2: Antifibrotic Effects of this compound on ECM Protein Synthesis in SSc Fibroblasts
| ECM Protein | Inhibition by this compound (in vitro) |
| Collagen 1a1 | Up to 90% |
| Collagen 1a2 | Up to 90% |
| Fibronectin-1 | Up to 90% |
Idiopathic Pulmonary Fibrosis Research
Idiopathic Pulmonary Fibrosis (IPF) is a severe fibrotic lung disease for which this compound has been investigated due to its antifibrotic properties ersnet.orgtandfonline.comsciencedaily.com. This compound is an intracellular inhibitor of multiple tyrosine kinases implicated in fibrogenic pathways relevant to IPF ersnet.orgatsjournals.org. Preclinical studies using mouse models of pulmonary fibrosis have demonstrated this compound mesylate's antifibrotic effects smw.chnih.govtandfonline.com.
However, the efficacy of this compound in human IPF patients has yielded different results. A randomized, placebo-controlled, 96-week trial involving 119 patients with IPF investigated the safety and clinical effects of this compound ersnet.orgatsjournals.orgatsjournals.org. The trial results indicated no significant benefits for this compound with respect to the primary endpoint, defined as time to disease progression (10% decline in percent predicted Forced Vital Capacity (FVC) from baseline) or time to death ersnet.orgatsjournals.orgatsjournals.org. There was no significant effect of this compound therapy on changes in FVC or diffusing capacity of carbon monoxide at 48, 72, or 96 weeks atsjournals.orgatsjournals.org. While a change in resting PaO2 favored this compound therapy at 48 weeks (P = 0.005), this effect was not sustained at 96 weeks (P = 0.074) atsjournals.orgatsjournals.org. Additionally, there was no significant difference in mortality between the this compound and placebo groups during the 96-week trial (8 deaths in this compound group vs. 10 in placebo group; log rank P = 0.64) atsjournals.orgatsjournals.org. Based on these findings, current clinical practice guidelines include a strong recommendation against the use of this compound in the treatment of IPF ersnet.org.
Table 3: this compound Clinical Trial Outcomes in Idiopathic Pulmonary Fibrosis (96 Weeks)
| Outcome Parameter | This compound Group (n=59) | Placebo Group (n=60) | Statistical Significance (P-value) |
| Time to disease progression/death | Not significant | Not significant | P = 0.89 ersnet.orgatsjournals.org |
| Change in FVC at 48, 72, 96 weeks | No effect | No effect | P > 0.39 at all time points atsjournals.org |
| Change in diffusing capacity of carbon monoxide | No effect | No effect | P > 0.26 at all time points atsjournals.org |
| Change in resting PaO2 at 48 weeks | Favored this compound | - | P = 0.005 atsjournals.org |
| Change in resting PaO2 at 96 weeks | Not significant | - | P = 0.074 atsjournals.org |
| Deaths (96 weeks) | 8 | 10 | P = 0.64 atsjournals.org |
Modulation of Fibroblast Proliferation and Migration
This compound has been shown to significantly inhibit the proliferation and migration of fibroblasts, which are critical processes in the development and progression of fibrosis. Studies have demonstrated that this compound dose-dependently inhibits the growth of lung fibroblasts and their DNA synthesis, particularly when stimulated by Platelet-Derived Growth Factor (PDGF) atsjournals.org. For instance, this compound treatment at concentrations as low as 0.1 to 0.3 µM effectively suppressed proliferative responses in NIH3T3 and primary lung fibroblasts stimulated by PDGF-AA and PDGF-BB atsjournals.org. However, it did not significantly affect proliferation stimulated by epidermal growth factor (EGF) or fibroblast growth factor 2 (FGF-2) atsjournals.org.
In human breast stromal fibroblasts, this compound inhibits proliferation by blocking the activation of the extracellular signal-regulated kinase (ERK) and Akt signaling pathways and by up-regulating the cyclin-dependent kinase inhibitor p21WAF1, leading to cell cycle arrest at the G0/G1 phase aacrjournals.orgnih.gov. The inhibition of PDGF-mediated fibroblast proliferation was found to be more potent and primarily due to the inhibition of the Akt pathway aacrjournals.orgnih.gov.
Furthermore, this compound has been observed to impair profibrogenic cytokine-induced human pulmonary fibroblast migration in vitro nih.gov. For example, 1 µM this compound reduced the percentage of migrated normal human pulmonary fibroblasts stimulated by EGF and PDGF-AB, and this reduction became more pronounced at higher concentrations (e.g., 51.6% reduction at 10 µM) nih.gov. This suggests that this compound acts as a potential and specific inhibitor of fibroblast accumulation in fibrotic conditions nih.gov. In addition, this compound treatment impaired the migration of both pericytes and fibroblasts, with PDGF-BB identified as a major serum factor promoting this migration researchgate.net.
Table 1: Effect of this compound on Fibroblast Proliferation and Migration
| Fibroblast Type / Model | Stimulus | This compound Concentration (µM) | Effect on Proliferation/Migration | Snippet Index |
| Primary murine lung fibroblasts | PDGF-AA, PDGF-BB | 0.1-0.3 | Dose-dependent inhibition | atsjournals.org |
| Human breast stromal fibroblasts | PDGF | 0.1-10 | Inhibition, G0/G1 arrest | aacrjournals.orgnih.gov |
| Human pulmonary fibroblasts | Profibrogeneic cytokines (EGF, PDGF-AB) | 1, 10 | Inhibition of migration | nih.gov |
| Pericytes and Fibroblasts (in vitro) | 10% FCS, PDGF-BB | 2 | Complete abrogation of migration | researchgate.net |
| SSc fibroblasts | Unstimulated / TGF-β | Not specified | Reduced basal collagen gene expression | nih.gov |
Myofibroblast Differentiation Pathways
Myofibroblasts are key effector cells in fibrosis, characterized by increased ECM production and contractile properties. This compound has demonstrated the ability to prevent the differentiation of resting fibroblasts into myofibroblasts nih.govuzh.ch. In preclinical models of systemic sclerosis (SSc), such as the tight skin 1 (TSK-1) mouse model, this compound treatment reduced dermal and hypodermal thickening and prevented this critical transition nih.govuzh.ch.
While this compound can suppress transforming growth factor-beta (TGF-β)-induced myofibroblast differentiation through SMAD-independent pathways when administered during the injury phase, some studies suggest that it might not significantly affect the stably differentiated myofibroblast phenotype or epithelial-mesenchymal transition (EMT) if administered during the post-injury reparation phase researchgate.net. This implies a temporal sensitivity to this compound's antifibrotic effects, being more effective in early fibrogenic processes researchgate.netnih.gov.
Preclinical studies have also shown that this compound treatment can lead to a decrease in type I collagen and fibronectin-1 synthesis by both dermal fibroblasts and myofibroblasts nih.govoup.com. This reduction in ECM protein synthesis is a crucial aspect of its antifibrotic action.
Table 2: this compound's Impact on Myofibroblast Differentiation and ECM Production
| Cell Type / Model | Effect of this compound | Key Pathway/Mechanism | Snippet Index |
| Resting fibroblasts (TSK-1 mice) | Prevention of differentiation into myofibroblasts | Inhibition of c-Abl and PDGFR pathways | nih.govuzh.ch |
| Dermal fibroblasts and myofibroblasts | Decreased Type I collagen and fibronectin-1 synthesis | Inhibition of c-Abl | nih.gov |
| SSc fibroblasts (in vitro) | >80% reduction in Type I collagen gene expression | c-Abl blockade | nih.gov |
| Rat cardiac fibroblasts | Reduced collagen type I and III mRNA expression | Inhibition of PDGFR-β and c-Abl phosphorylation | ahajournals.org |
| Breast stromal fibroblasts | Inhibited collagen accumulation (collagen I, III mRNA downregulation) | Down-regulation of mRNA synthesis of collagen I and III | aacrjournals.orgnih.gov |
Inhibition of c-Abl and PDGFR Tyrosine Kinases in Fibrogenesis
A cornerstone of this compound's antifibrotic mechanism lies in its potent and specific inhibition of c-Abl (Abelson kinase) and Platelet-Derived Growth Factor Receptor (PDGFR) tyrosine kinases atsjournals.orgnih.govnih.govahajournals.orgsmw.chersnet.orgatsjournals.orgnih.govresearchgate.nettandfonline.com. These tyrosine kinases play central roles in promoting fibrogenesis.
The c-Abl kinase, a non-receptor tyrosine kinase, is a significant downstream target of pro-fibrotic Transforming Growth Factor-beta (TGF-β) signaling oup.comsmw.ch. TGF-β is a central mediator in pro-fibrotic diseases, and its induction of extracellular matrix proteins is markedly reduced in cells deficient for c-Abl oup.comsmw.ch. This compound effectively blocks the tyrosine kinase activity of c-Abl, thereby interfering with TGF-β's pro-fibrotic effects nih.govnih.govoup.comahajournals.orgsmw.chnih.gov. For example, in experimental models, this compound abrogated the TGF-β-induced stimulation of Type I collagen production and other fibrotic gene products like fibronectin, connective tissue growth factor, and plasminogen activator inhibitor-1 nih.gov. It also reduced basal collagen gene expression in SSc fibroblasts nih.gov.
Simultaneously, this compound suppresses the tyrosine kinase activity of the PDGFR atsjournals.orgnih.govnih.govahajournals.orgsmw.chersnet.orgatsjournals.orgnih.govresearchgate.nettandfonline.com. PDGF can directly stimulate fibroblasts to contract collagen matrices and differentiate into myofibroblasts, and it also upregulates the expression of TGF-β1 and c-Abl in fibroblasts ahajournals.org. By inhibiting PDGFR, this compound blocks a major pro-fibrotic pathway that stimulates fibrogenesis and enhances fibroblast contractility smw.ch. Studies in rat cardiac fibroblasts have shown that this compound reduces collagen production by inhibiting the phosphorylation of both c-Abl and PDGFR-β, particularly under co-incubation with PDGF-BB and TGF-β1 ahajournals.org.
The combined inhibition of c-Abl and PDGFR by this compound is considered critical for its antifibrotic effects uzh.chersnet.orgresearchgate.net. This dual targeting mechanism allows this compound to disrupt multiple interdependent pathways that drive fibrosis, including the production of extracellular matrix and the contractility of fibroblasts oup.comsmw.ch. In models of radiation-induced fibrosis, this compound not only reduced the activation of PDGFR-β and c-Abl but also decreased the levels of TGF-β, suggesting an interruption of cross-talk between these pathways nih.gov.
Table 3: Key Tyrosine Kinase Targets of this compound in Fibrogenesis
| Tyrosine Kinase Target | Role in Fibrogenesis | This compound's Effect | Snippet Index |
| c-Abl | Downstream target of TGF-β; promotes fibrosis, fibroblast growth, matrix gene expression | Inhibited activity; prevents TGF-β induced ECM production | nih.govnih.govoup.comahajournals.orgsmw.chnih.govtandfonline.com |
| PDGFR | Stimulates fibrogenesis, enhances fibroblast contractility, promotes ECM production | Inhibited activity; reduces fibroblast proliferation and migration, decreases collagen synthesis | atsjournals.orgnih.govresearchgate.netnih.govahajournals.orgsmw.chersnet.orgatsjournals.orgnih.govresearchgate.nettandfonline.com |
| c-Kit | Implicated in pulmonary fibrosis | Inhibited activity | atsjournals.orgnih.goversnet.orgatsjournals.org |
Advanced Methodologies in Imatinib Research
Proteomics Approaches for Target Identification
Proteomics, the large-scale study of proteins, offers powerful tools to identify the direct and indirect cellular targets of imatinib, as well as to quantify changes in protein expression and phosphorylation states induced by the drug.
Affinity Purification with Immobilized Kinase Inhibitors
Affinity purification, particularly using immobilized kinase inhibitors, is a prominent chemical proteomics strategy to identify direct cellular targets of kinase-selective drugs like this compound. This method involves using a resin to which non-selective kinase inhibitors are covalently attached. When cell lysates are passed over this resin, protein kinases that can bind to the inhibitors are retained. By comparing the proteins bound to the resin from untreated cells versus this compound-treated cells, researchers can identify kinases whose binding is affected by this compound, thus revealing potential direct targets or off-targets.
A proof-of-concept proteomics study applied this strategy to chronic myeloid leukemia cells expressing the Bcr-Abl fusion kinase. This integrated approach indicated additional this compound target candidates, such as flavine adenine dinucleotide synthetase, as well as repressed phosphorylation events on downstream effectors not yet implicated in this compound-regulated signaling. These included activity-regulating phosphorylations on the kinases Btk, Fer, and focal adhesion kinase, which may qualify them as alternative target candidates in Bcr-Abl-driven oncogenesis.
SILAC-based Quantitative Proteomics
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) is a robust quantitative proteomics technique widely used to measure global protein expression changes and identify perturbed biological pathways in response to drug treatment. In SILAC, cells are metabolically labeled with "light" or "heavy" isotopic forms of amino acids, allowing for the precise quantification of protein abundance differences between treated and untreated samples via mass spectrometry.
SILAC has been instrumental in dissecting this compound's effects on the proteome of K562 human chronic myeloid leukemia cells, which express the Bcr-Abl fusion kinase. For instance, studies have shown that this compound treatment leads to significant alterations in the expression levels of numerous proteins. One study quantified 1344 proteins, finding 73 that had significantly altered levels of expression induced by this compound and could be quantified in both forward and reverse SILAC labeling experiments. These included the down-regulation of thymidylate synthase, S-adenosylmethionine synthetase, and glycerol-3-phosphate dehydrogenase, as well as the up-regulation of poly(ADP-ribose) polymerase 1, hemoglobins, and enzymes involved in heme biosynthesis. Furthermore, this compound-induced hemoglobinization and erythroid differentiation in K562 cells were found to be associated with global histone H4 hyperacetylation. These results provided potential biomarkers for monitoring therapeutic intervention of CML using this compound and offered new knowledge for gaining insight into its molecular mechanisms of action.
Another study utilized SILAC to compare proteome changes induced by this compound and novel this compound derivatives in K562 cells, quantifying a total of 986 proteins. This approach allowed for the identification of significantly altered proteins upon treatment with these compounds.
Table 1: Examples of Proteins with Altered Expression or Binding in K562 Cells Upon this compound Treatment (Proteomics Approaches)
| Protein Name | Regulation by this compound (Observed Effect) | Methodology | Source |
| Flavine Adenine Dinucleotide Synthetase | Candidate Target (Binding Alteration) | Affinity Purification | |
| Btk | Repressed Phosphorylation Events | Affinity Purification | |
| Fer | Repressed Phosphorylation Events | Affinity Purification | |
| Focal Adhesion Kinase | Repressed Phosphorylation Events | Affinity Purification | |
| Thymidylate Synthase | Down-regulation of expression | SILAC Quantitative Proteomics | |
| S-adenosylmethionine Synthetase | Down-regulation of expression | SILAC Quantitative Proteomics | |
| Glycerol-3-phosphate Dehydrogenase | Down-regulation of expression | SILAC Quantitative Proteomics | |
| Poly(ADP-ribose) Polymerase 1 | Up-regulation of expression | SILAC Quantitative Proteomics | |
| Hemoglobins | Up-regulation of expression | SILAC Quantitative Proteomics | |
| NHERF1 (Na+/H+ exchanger regulatory factor 1) | Strongly upregulated (in this compound-resistant cells) | 2-DE-MS Proteomics | |
| Histone H4 | Hyperacetylation | SILAC Quantitative Proteomics |
Bioinformatics and Systems Biology Approaches
Bioinformatics and systems biology approaches are essential for integrating large-scale biological data, such as gene expression profiles and proteomic datasets, to construct networks and models that provide a holistic understanding of this compound's effects and the underlying disease dynamics.
Gene Expression Profiling and Network Analysis (e.g., Weighted Gene Co-expression Network Analysis (WGCNA))
Gene expression profiling, often performed using microarray or RNA sequencing, allows for the identification of differentially expressed genes (DEGs) in response to this compound treatment or in this compound-resistant cells. Subsequent network analysis, including Weighted Gene Co-expression Network Analysis (WGCNA), helps to uncover modules of highly correlated genes and identify hub genes that may play crucial roles in disease progression or drug response.
In the context of this compound research, gene expression profiling has revealed important insights into resistance mechanisms and disease pathways. Studies comparing this compound-sensitive and this compound-resistant K562 cells have identified hundreds of DEGs. Pathway enrichment analyses of these DEGs have highlighted their involvement in processes such as cell adhesion, cell migration, differentiation, inflammatory response, and signaling pathways like Rap1, focal adhesion, and those regulating pluripotency of stem cells.
WGCNA has been applied to analyze gene expression profiles from this compound-treated CML patients to identify immune-related genes (IRGs) associated with treatment response. This analysis categorized patients into high- and low-immune score groups and identified 428 differentially expressed IRGs. Functional enrichment analysis revealed that these genes were involved in immune-related pathways, including T-cell receptor signaling and cytokine-cytokine receptor interaction. Based on five modules in WGCNA and top-ranked degree, 10 hub genes were identified, with IL10RA, SCN9A, and SLC26A11 identified as potential biomarkers for predicting this compound response. For instance, high expression of IL10RA and SLC26A11, and low expression of SCN9A, were observed in responders.
WGCNA has also been used to explore this compound's effects in other conditions, such as rheumatoid arthritis, where it helped identify CSF1R as a core gene involved in inflammation that this compound could mitigate by suppressing its expression.
Mathematical Modeling of Immune Response Dynamics
Mathematical modeling plays a crucial role in understanding the complex dynamics of the immune system in chronic myeloid leukemia (CML) and its interaction with this compound therapy. These models often employ systems of differential equations to simulate the interplay between healthy cells, leukemic cells, and various immune cell populations.
Early mathematical models of CML dynamics under this compound treatment often did not incorporate the anti-leukemia immune response. However, recent experimental data indicating that this compound treatment may promote the development of anti-leukemia immune responses as patients enter remission have led to the development of more comprehensive models. These models, often utilizing delay differential equations to account for the duration of cell division, suggest that anti-leukemia T cell responses may play a critical role in maintaining CML patients in remission under this compound therapy. Furthermore, models propose that this compound therapy might drive leukemic cell populations to enter and fall below an "optimal load zone" too rapidly to sustain the anti-leukemia T cell response, suggesting vaccination approaches in combination with this compound could help sustain this response for a durable cure.
More recent mathematical models have delved into the intricacies of this compound's impact on specific T cell subsets, such as T helper (Th) cells and Treg cells, and their potential role in allergic reactions induced by the drug. These models integrate cellular interactions, drug pharmacokinetics, and immune responses to unveil mechanisms underlying the dominance of Th2 over Th1 and Treg cells, leading to allergic manifestations. Such models can also explore how this compound affects the maturation and functioning of Antigen-Presenting Cells (APCs) and, consequently, T cells within the immune system.
Table 2: Key Cellular Populations and Interactions Modeled in this compound-Immune Dynamics
| Cellular Population/Process | Description | Relevance to this compound Research (as per models) | Source |
| Leukemic Cells | Abnormal proliferation in CML (e.g., Bcr-Abl positive cells) | Direct target of this compound; mathematical models track their reduction. | |
| Healthy Hematopoietic Cells | Normal blood cell production | Modeled in relation to leukemic cell suppression and immune interactions. | |
| Anti-leukemia T Cells | Immune cells targeting and killing leukemic cells | Their persistence is crucial for maintaining remission under this compound therapy. | |
| T Helper (Th) Cells | Orchestrate immune responses, differentiate into Th1/Th2. | This compound's impact on their subsets (Th1, Th2, Treg) is modeled, particularly regarding allergic reactions. | |
| Regulatory T (Treg) Cells | Suppress immune responses to maintain self-tolerance. | Modeled concerning this compound-induced alterations and allergic manifestations. | |
| Antigen-Presenting Cells (APCs) | Process and present antigens to T cells. | This compound is considered to modulate their maturation and proper functioning. | |
| Drug Pharmacokinetics | Absorption, distribution, metabolism, excretion of this compound. | Integrated into models to link drug concentration to cellular and immune effects. |
Molecular Docking and Computational Chemistry for Derivative Design
Molecular docking and computational
Q & A
Q. How should researchers design preclinical experiments to evaluate Imatinib’s efficacy in chronic myeloid leukemia (CML)?
- Methodological Answer : Begin with in vitro dose-response assays using BCR-ABL+ cell lines, referencing prior studies that established IC50 values (e.g., 0.25–1.0 µM) . Validate findings with in vivo models (e.g., xenografts) using doses of 50–100 mg/kg/day, adjusted for bioavailability . Include controls for off-target effects (e.g., ABL-negative cell lines) and replicate experiments at least three times to ensure statistical power . For translational relevance, cross-reference clinical trial data on plasma trough levels (e.g., ≥1,000 ng/mL correlates with cytogenetic response) .
Q. What factors should be controlled to ensure reliable this compound response data in cell-based assays?
- Methodological Answer : Control for P-glycoprotein (P-gp) expression, which mediates this compound efflux and reduces intracellular drug accumulation . Use flow cytometry to quantify P-gp levels in cell lines and correlate with IC50 shifts. Standardize culture conditions (e.g., serum concentration, pH) to minimize variability in proliferation rates. Include a positive control (e.g., STI571-resistant cell lines with KIT mutations) and validate results with secondary assays (e.g., apoptosis via Annexin V staining) .
Q. How can researchers optimize this compound dosing in early-phase clinical trials?
- Methodological Answer : Use pharmacokinetic/pharmacodynamic (PK/PD) modeling to link plasma trough levels (target: 1,000–3,200 ng/mL) to clinical outcomes . Adjust doses based on patient-specific factors (e.g., hepatic function, drug-drug interactions) and monitor adverse events (e.g., edema, myelosuppression) . For dose escalation, follow phase I protocols with cohorts receiving 25–1,000 mg/day, prioritizing safety and response rates .
Advanced Research Questions
Q. How should researchers analyze contradictory data on this compound’s efficacy in non-CML malignancies (e.g., gastrointestinal stromal tumors [GIST])?
- Methodological Answer : Conduct subgroup analyses stratified by tumor genotype (e.g., KIT exon 11 vs. PDGFRA mutations) . Use multivariate Cox regression to identify confounding variables (e.g., mitotic index ≤5/50 HPFs correlates with prolonged progression-free survival) . Cross-validate findings with independent datasets (e.g., NCT00075426 trial data) and employ sensitivity analyses to assess robustness against outliers .
Q. What experimental strategies can elucidate mechanisms of this compound resistance in advanced CML?
- Methodological Answer : Perform Sanger sequencing of BCR-ABL kinase domains to detect resistance-conferring mutations (e.g., T315I) . Combine in vitro mutagenesis screens with structural modeling to predict mutation impact on drug binding . Validate findings using patient-derived xenografts (PDXs) and correlate with clinical resistance timelines. Explore adjunct therapies (e.g., dasatinib for T315I mutations) .
Q. How can circulating tumor DNA (ctDNA) be utilized to assess minimal residual disease (MRD) in GIST patients discontinuing this compound?
- Methodological Answer : Design longitudinal studies with serial ctDNA sampling during this compound interruption. Use digital PCR or NGS to detect KIT/PDGFRA mutations, with a sensitivity threshold of 0.1% variant allele frequency . Correlate ctDNA dynamics with radiographic progression (RECIST 1.1) and survival outcomes. Note: Current ctDNA platforms may miss non-KIT/PDGFRA mutations, necessitating orthogonal validation (e.g., ddPCR) .
Q. What statistical methods are appropriate for analyzing survival outcomes in this compound interruption trials?
- Methodological Answer : Apply Kaplan-Meier estimates for progression-free survival (PFS) and log-rank tests to compare groups (e.g., complete vs. incomplete tumor resection) . Use Cox proportional hazards models to adjust for covariates (e.g., peritoneal metastasis status, mitotic index) . For small cohorts, employ bootstrapping to estimate confidence intervals and address censoring biases .
Methodological and Ethical Considerations
Q. How should researchers address retracted or conflicting studies in meta-analyses of this compound data?
- Methodological Answer : Exclude retracted papers (e.g., ) after verifying retraction notices in databases like Retraction Watch. Use GRADE criteria to assess study quality and heterogeneity (e.g., I² statistic). For conflicting results, perform sensitivity analyses by excluding outlier studies and report funnel plots to detect publication bias .
Q. What guidelines should govern the compilation of this compound data in Investigator’s Brochures (IBs) for clinical trials?
- Methodological Answer : Follow ICH E6(R2) guidelines: Include pharmacokinetic data (e.g., Cmax, AUC), toxicity profiles, and response rates from phase I-III trials . For novel formulations (e.g., generics), provide bioequivalence data vs. the reference product. Update IBs annually or after significant safety events (e.g., new black-box warnings) .
Q. How can researchers ethically design trials involving this compound interruption in stable GIST patients?
- Methodological Answer :
Obtain informed consent detailing risks of disease progression (61% in 19.6 months) and benefits of re-introduction (88.6% response rate) . Use a Data Safety Monitoring Board (DSMB) to review interim analyses. Ensure rescue protocols (e.g., this compound re-initiation at 400 mg/day) are predefined in the trial protocol .
Retrosynthesis Analysis
AI-Powered Synthesis Planning: Our tool employs the Template_relevance Pistachio, Template_relevance Bkms_metabolic, Template_relevance Pistachio_ringbreaker, Template_relevance Reaxys, Template_relevance Reaxys_biocatalysis model, leveraging a vast database of chemical reactions to predict feasible synthetic routes.
One-Step Synthesis Focus: Specifically designed for one-step synthesis, it provides concise and direct routes for your target compounds, streamlining the synthesis process.
Accurate Predictions: Utilizing the extensive PISTACHIO, BKMS_METABOLIC, PISTACHIO_RINGBREAKER, REAXYS, REAXYS_BIOCATALYSIS database, our tool offers high-accuracy predictions, reflecting the latest in chemical research and data.
Strategy Settings
| Precursor scoring | Relevance Heuristic |
|---|---|
| Min. plausibility | 0.01 |
| Model | Template_relevance |
| Template Set | Pistachio/Bkms_metabolic/Pistachio_ringbreaker/Reaxys/Reaxys_biocatalysis |
| Top-N result to add to graph | 6 |
Feasible Synthetic Routes




Featured Recommendations
| Most viewed |
|
|
|---|---|---|
| Most popular with customers |
|
Disclaimer and Information on In-Vitro Research Products
Please be aware that all articles and product information presented on BenchChem are intended solely for informational purposes. The products available for purchase on BenchChem are specifically designed for in-vitro studies, which are conducted outside of living organisms. In-vitro studies, derived from the Latin term "in glass," involve experiments performed in controlled laboratory settings using cells or tissues. It is important to note that these products are not categorized as medicines or drugs, and they have not received approval from the FDA for the prevention, treatment, or cure of any medical condition, ailment, or disease. We must emphasize that any form of bodily introduction of these products into humans or animals is strictly prohibited by law. It is essential to adhere to these guidelines to ensure compliance with legal and ethical standards in research and experimentation.
