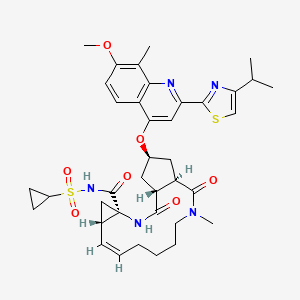
Simeprevir
Description
This compound is an oral, direct acting hepatitis C virus (HCV) protease inhibitor that is no longer available but was previously used in combination with other antiviral agents in the treatment of chronic hepatitis C, genotypes 1 and 4, but subsequently was withdrawn. This compound has been linked to isolated, rare instances of mild, acute liver injury during treatment and to occasional cases of hepatic decompensation in patients with preexisting cirrhosis.
This compound is an orally bioavailable inhibitor of the hepatitis C virus (HCV) protease complex comprised of non-structural protein 3 and 4A (NS3/NS4A), with activity against HCV genotype 1. Upon administration, this compound reversibly binds to the active center and binding site of the HCV NS3/NS4A protease and prevents NS3/NS4A protease-mediated polyprotein maturation. This disrupts both the processing of viral proteins and the formation of the viral replication complex, which inhibits viral replication in HCV genotype 1-infected host cells. NS3, a serine protease, is essential for the proteolytic cleavage of multiple sites within the HCV polyprotein and plays a key role during HCV ribonucleic acid (RNA) replication. NS4A is an activating factor for NS3. HCV is a small, enveloped, single-stranded RNA virus belonging to the Flaviviridae family; HCV infection is associated with the development of hepatocellular carcinoma (HCC).
Oral HCV-PROTEASE INHIBITOR effective against hepatitis C virus (HCV) serine protease NS3/4A. It is used in the treatment of chronic hepatitis C (Antivirals) genotype 1 infection in adults with compensated liver disease, including CIRRHOSIS.
Properties
Key on ui mechanism of action |
Simeprevir is accumulated in the liver after uptake into hepatocytes via OATP1B1/3. NS3/4A heterodimeric complex is composed of the cofactor N4A subunit and N3 subunit which contains the proteolytic site. The NS3/4A protease cleaves the HCV polyprotein downstream of the NS3 site, generating non-structural viral proteins NS3, NS4A, NS4B, NS5A and NS5B and subsequently formation of mature proteins. Simeprevir exerts an inhibitory action on HCV polyprotein cleavage via induced-fit binding to an extended S2 subsite located in the NS3 catalytic site. NS3/4A inhibitors usually depend on few interactions located in the substrate binding groove of the viral serine protease, thus are susceptible to resistance and failed treatment from few critical mutations in these sites. At higher concentration above their antiviral half-maximal effective concentration (EC50), simeprevir and other NS3/4A inhibitors also restore interferon (IFN)-signaling pathways that are thought to be disrupted by NS3/4A protease and recover innate immune processes. NS3/4A protease cleaves two essential adaptor proteins that initiate signaling leading to activation of IFN regulatory factor 3 and IFN-α/β synthesis, which are mitochondrial antiviral-signaling proteins (MAVS otherwise known as IPS-1, VISA, or Cardif) and toll/interleukin-1 receptor (TIR)- domain-containing adaptor-inducing IFN-β (TRIF). Blocking the function of these adaptor proteins results in impaired interferon induction. NS3/4A inhibitors recover the proper IFN-signaling pathways. Simeprevir sodium is a selective hepatitis C virus (HCV) nonstructural 3/4A (NS3/4A) protease inhibitor (PI). The drug is a direct-acting antiviral (DAA) with activity against HCV. Simeprevir binds noncovalently to the active site of HCV NS3/4A protease, thereby blocking enzyme activity essential for viral replication (i.e., cleavage of the HCV-encoded polyprotein at the NS3/NS4A, NS4A/NS4B, NS4A/NS5A, and NS5A/NS5B junctions). In vitro studies using biochemical and cell-based replicon assays indicate that simeprevir has potent activity against HCV genotypes 1a and 1b1 5 8 9 and has some activity against HCV genotypes 2, 4, 5, and 6 (not genotype 3). |
|---|---|
CAS No. |
923604-59-5 |
Molecular Formula |
C38H47N5O7S2 |
Molecular Weight |
749.9 g/mol |
IUPAC Name |
(4S)-N-cyclopropylsulfonyl-17-[7-methoxy-8-methyl-2-(4-propan-2-yl-1,3-thiazol-2-yl)quinolin-4-yl]oxy-13-methyl-2,14-dioxo-3,13-diazatricyclo[13.3.0.04,6]octadec-7-ene-4-carboxamide |
InChI |
InChI=1S/C38H47N5O7S2/c1-21(2)30-20-51-35(40-30)29-18-32(26-13-14-31(49-5)22(3)33(26)39-29)50-24-16-27-28(17-24)36(45)43(4)15-9-7-6-8-10-23-19-38(23,41-34(27)44)37(46)42-52(47,48)25-11-12-25/h8,10,13-14,18,20-21,23-25,27-28H,6-7,9,11-12,15-17,19H2,1-5H3,(H,41,44)(H,42,46)/t23?,24?,27?,28?,38-/m0/s1 |
InChI Key |
JTZZSQYMACOLNN-ZMKZURCUSA-N |
SMILES |
CC1=C(C=CC2=C1N=C(C=C2OC3CC4C(C3)C(=O)N(CCCCC=CC5CC5(NC4=O)C(=O)NS(=O)(=O)C6CC6)C)C7=NC(=CS7)C(C)C)OC |
Isomeric SMILES |
CC1=C(C=CC2=C1N=C(C=C2OC3CC4C(C3)C(=O)N(CCCCC=CC5C[C@@]5(NC4=O)C(=O)NS(=O)(=O)C6CC6)C)C7=NC(=CS7)C(C)C)OC |
Canonical SMILES |
CC1=C(C=CC2=C1N=C(C=C2OC3CC4C(C3)C(=O)N(CCCCC=CC5CC5(NC4=O)C(=O)NS(=O)(=O)C6CC6)C)C7=NC(=CS7)C(C)C)OC |
Color/Form |
White to almost white powder |
Pictograms |
Irritant; Environmental Hazard |
solubility |
Insoluble Practically insoluble in water over a wide pH range Practically insoluble in propylene glycol; very slightly soluble in ethanol; slightly soluble in acetone. Soluble in dichloromethane; freely soluble in some organic solvents |
Synonyms |
435, TMC 435350, TMC N-(17-(2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinolin-4-yloxy)-13-methyl-2,14-dioxo-3,13-diazatricyclo(13.3.0.04,6)octadec-7-ene-4-carbonyl)(cyclopropyl)sulfonamide Olysio simeprevir TMC 435 TMC 435350 TMC-435 TMC-435350 TMC435 TMC435350 |
vapor_pressure |
5.9X10-27 mm Hg at 25 °C (est) |
Origin of Product |
United States |
Molecular Mechanisms of Simeprevir's Antiviral Activity
Inhibition of Hepatitis C Virus (HCV) NS3/4A Protease Function
Simeprevir functions as a highly specific inhibitor of the Hepatitis C Virus NS3/4A protease, a critical enzyme for the viral life cycle. drugbank.compatsnap.comnih.govdrugs.comnih.govdovepress.comselleckchem.comnih.gov This protease is a serine protease, meaning it utilizes a serine residue in its active site for catalysis, and it is essential for the replication of HCV. drugbank.compatsnap.comnih.govdrugs.comdovepress.comnih.govnih.govasm.org
The Hepatitis C Virus genome encodes a large single polyprotein precursor, which must be precisely cleaved into ten individual, functional proteins by both host and viral proteases to enable viral replication. drugbank.compatsnap.comdovepress.comnih.govnih.govnih.gov The NS3/4A protease is specifically responsible for cleaving this polyprotein at several sites, leading to the generation of mature non-structural viral proteins such as NS3, NS4A, NS4B, NS5A, and NS5B. These mature proteins are vital for the formation of the viral replication complex and subsequent viral assembly. drugbank.compatsnap.comdovepress.comnih.govnih.govasm.orgnih.govprobes-drugs.org By inhibiting the NS3/4A protease, this compound effectively prevents this crucial polyprotein cleavage. drugbank.compatsnap.comdrugs.comnih.govasm.orgpatsnap.com This disruption halts the production of functional viral proteins, thereby blocking the viral replication process early in the HCV life cycle and significantly reducing the viral load. drugbank.compatsnap.comdrugs.comnih.govpatsnap.com
This compound is categorized as a second-generation HCV protease inhibitor, distinguishing it from earlier compounds like Boceprevir and Telaprevir. drugbank.comnih.govnih.govdovepress.comnih.govresearchgate.net A key characteristic of this compound's interaction with its target is its competitive, reversible, and noncovalent binding modality. drugbank.comnih.govnih.govselleckchem.comnih.govnih.govresearchgate.net Its macrocyclic structure plays a significant role in enhancing its affinity and selectivity for the NS3 protease. This structural feature facilitates rapid association with the enzyme and a slow dissociation rate, contributing to its potent inhibitory effect through noncovalent interactions. drugbank.comnih.govnih.govdovepress.com
This compound exerts its inhibitory action on the HCV NS3/4A protease through an induced-fit binding mechanism. drugbank.comnih.govprobes-drugs.orgnih.gov This involves the drug binding to an extended S2 subsite located within the NS3 catalytic site. drugbank.comnih.govprobes-drugs.orgnih.gov The binding of inhibitors, including this compound, can induce conformational changes not only in the immediate vicinity of the active site but also in more distant amino acid residues of the enzyme. This inherent flexibility of the induced-fit mechanism is crucial for the drug's interaction but also presents challenges, as mutations in the viral protease can affect this favorable binding and contribute to drug resistance.
Competitive, Reversible, and Noncovalent Binding Modalities
Modulation of Host Innate Immune Signaling Pathways
Beyond its direct antiviral action, this compound also influences host innate immune responses. The HCV NS3/4A protease is known to interfere with the host's innate immune system, a critical first line of defense against viral infections. dovepress.comnih.govasm.orgprobes-drugs.orgmedcraveonline.comfrontiersin.orgwjgnet.com Specifically, the NS3/4A protease cleaves key cellular adaptor proteins, such as mitochondrial antiviral-signaling proteins (MAVS, also known as IPS-1, VISA, or Cardif) and Toll/interleukin-1 receptor (TIR)-domain-containing adaptor-inducing IFN-β (TRIF). drugbank.comnih.govasm.orgnih.govprobes-drugs.orgfrontiersin.orgwjgnet.com This cleavage disrupts the signaling cascades that would normally lead to the activation of interferon regulatory factor 3 (IRF3) and the subsequent production of type I interferons (IFN-α/β) and type III interferons (IFN-λ), which are essential for initiating antiviral responses. drugbank.comnih.govasm.orgnih.govprobes-drugs.orgfrontiersin.orgmdpi.comnih.gov
Synergistic and Additive Antiviral Effects in In Vitro Models
This compound has demonstrated beneficial interactions when combined with other antiviral agents in in vitro models. In HCV replicon cells, this compound has shown synergistic effects when combined with interferon-α and HCV NS5B inhibitors, such as Sofosbuvir. drugbank.compatsnap.comdovepress.comnih.gov Additionally, it exhibits additive antiviral effects with ribavirin in these models. drugbank.compatsnap.comnih.gov
Beyond its primary indication for HCV, this compound has also shown broader antiviral potential. Research has indicated that this compound can synergize with remdesivir in suppressing the replication of SARS-CoV-2 in vitro. researchgate.netnih.govacs.orgbiorxiv.orgresearchgate.net This synergistic effect allows for potentially lower effective doses of remdesivir. nih.govbiorxiv.orgresearchgate.net The mechanism behind this observed synergy is attributed to this compound's unexpected inhibitory activity against both the SARS-CoV-2 Main protease (Mpro) and RNA-dependent RNA polymerase (RdRp) activities. nih.govacs.orgbiorxiv.orgresearchgate.net
Interactions with Interferon-alpha and HCV NS5B Inhibitors
This compound exhibits synergistic antiviral effects when combined with interferon-alpha (IFN-α) and HCV NS5B inhibitors in HCV replicon cell models. wikipedia.orgresearchgate.net This synergistic interaction underscores the potential for enhanced viral suppression when these agents are used in combination.
The NS3/4A protease, which this compound targets, is known to interfere with host interferon-signaling pathways. By inhibiting this viral protease, this compound can contribute to the recovery of proper IFN-signaling, thereby augmenting the host's innate immune response against HCV. wikipedia.org
Clinical studies have frequently evaluated this compound in combination regimens that include peginterferon alfa, a modified form of interferon-alpha. The addition of this compound to peginterferon alfa and ribavirin has demonstrated significantly improved sustained virological response (SVR) rates in patients with HCV genotype 1 infection compared to peginterferon alfa and ribavirin alone. For instance, in treatment-naïve patients with HCV genotype 1, studies like QUEST-1 and QUEST-2 reported SVR12 rates of approximately 80-81% with this compound in combination with peginterferon alfa 2a and ribavirin, compared to around 50% in the placebo group receiving peginterferon alfa and ribavirin alone.
HCV NS5B inhibitors, such as sofosbuvir, target the viral RNA-dependent RNA polymerase, another critical enzyme for HCV replication. The combination of this compound (an NS3/4A protease inhibitor) and sofosbuvir (an NS5B polymerase inhibitor) has shown high rates of sustained virological response, even in difficult-to-treat populations like patients with cirrhosis. Research has indicated that this compound in combination with an NS5B inhibitor can lead to synergistic antiviral activity. wikipedia.orgresearchgate.net
Interactions with Ribavirin
This compound demonstrates additive effects when combined with ribavirin in HCV replicon cells. wikipedia.orgresearchgate.net Ribavirin is a broad-spectrum antiviral nucleoside analog that inhibits viral genome synthesis, primarily by inhibiting inosine monophosphate dehydrogenase (IMP), an enzyme involved in GTP biosynthesis.
Ribavirin has been a long-standing component of hepatitis C treatment regimens, often used in combination with peginterferon alfa. mims.com The inclusion of this compound in triple therapy regimens with peginterferon alfa and ribavirin has been a common approach for treating chronic HCV infection, particularly for genotypes 1 and 4. This triple therapy has been shown to significantly improve SVR rates and allow for shorter treatment durations compared to dual therapy with peginterferon and ribavirin alone.
A meta-analysis of randomized controlled trials involving 2,301 patients demonstrated that the triple regimen of this compound, peginterferon, and ribavirin resulted in a higher pooled SVR rate and a lower pooled relapse rate compared to dual therapy.
Table 1: Research Findings on this compound Combination Therapies
| Combination Regimen | HCV Genotype(s) | Effect in Replicon Cells | Clinical Outcome (SVR12/SVR24) | References |
| This compound + Interferon-alpha + HCV NS5B Inhibitor | 1 | Synergistic | High SVR rates (e.g., with sofosbuvir) | wikipedia.orgresearchgate.net |
| This compound + Ribavirin | 1, 4 | Additive | Improved SVR rates in triple therapy (with peginterferon alfa) | wikipedia.orgresearchgate.net |
Structural Biology and Protein-ligand Interactions of Simeprevir
Crystallographic Analysis of Simeprevir-Bound HCV NS3/4A Protease Complexes
Intermolecular Interactions and Hydrogen Bonding Network
Role of K136 in Active Site Closure
Within the active site of the HCV NS3/4A protease, Lysine 136 (K136) plays a crucial role in the enzyme's catalytic mechanism and its interaction with inhibitors like this compound. During the enzymatic process, particularly in the transition state, the negatively charged oxygen (oxyanion) of the carboxylate tetrahedral intermediate is stabilized by hydrogen bonds with the backbone amides of Serine 138 (S138) and Glycine 137 (G137), as well as the side chain of K136. This interaction is vital for the stabilization of the oxyanion hole, a key feature in serine protease catalysis.
This compound has been shown to interact with K136 in crystal structures, including those of HCV genotypes 1b and 4a, highlighting its contribution to the inhibitor's binding pattern guidetopharmacology.org. While acidic side chains of natural substrates commonly form hydrogen bonds with K136, this interaction is less prevalent with inhibitors mims.com. Computational studies further emphasize the crucial, albeit distinct, contributions of K136 and S139 sidechains to the binding of HCV NS3/4A inhibitors. K136's ability to adopt different conformations in various crystal structures suggests a dynamic role in facilitating active site closure and optimal inhibitor binding mims.com.
Structure-Activity Relationship (SAR) Studies of this compound Derivatives
Extensive SAR studies were conducted during the development of this compound to understand how modifications to its chemical structure influence its potency, affinity, and selectivity against the HCV NS3/4A protease.
Influence of Macrocyclic Core on Affinity and Selectivity
The macrocyclic core is a defining feature of this compound, distinguishing it from first-generation linear peptidomimetic inhibitors wikidata.org. This macrocyclic portion significantly enhances the affinity and selectivity characteristics of the compound, enabling rapid association with and slow dissociation from the protein target through noncovalent binding wikidata.org. Macrocyclic structures generally offer functional diversity and stereochemical complexity in a semi-rigid, preorganized conformation, allowing them to bind with higher affinity and selectivity to challenging drug targets compared to more traditional small molecules.
This compound is a 14-membered P1-P3 macrocyclic inhibitor, developed through the strategic replacement of a hydroxyproline moiety with a cyclopentane surrogate within the peptidic framework. Studies involving the incorporation of a P1-P3 macrocyclization strategy and the examination of various ring sizes demonstrated that a 14-membered ring size was optimal for activity. This optimality underscores the critical interplay between the macrocycle and other substituents, such as those in the P3/P4 region. In certain cases, macrocyclic analogs have demonstrated significantly greater potency as NS3 protease inhibitors compared to their non-macrocyclic counterparts, with some macrocyclic derivatives being seven times more potent.
Role of Cyclopropylsulfonamide Moiety
The cyclopropylsulfonamide moiety is an integral part of the this compound structure, as indicated by its chemical name, which includes "N-(cyclopropylsulfonyl)". In the context of HCV NS3/4A protease inhibitors, acyl sulfonamide groups, which are structurally related to cyclopropylsulfonamide, have been shown to significantly improve potency. For instance, in related macrocyclic inhibitors, the replacement of a carboxylic acid group at P1 with an acylsulfonamide bioisostere led to over 100-fold improvement in potency in both enzyme and replicon assays. This highlights the critical contribution of the sulfonamide functionality to the inhibitory activity, likely through favorable interactions within the enzyme's active site.
Significance of Phenanthridine Ring
It is important to note that this compound's chemical structure contains a quinoline ring, specifically a "4-quinolinyl" group, rather than a phenanthridine ring. While phenanthridine moieties are present in some other HCV NS3/4A protease inhibitors, such as paritaprevir mims.com, the significance of a phenanthridine ring cannot be discussed in relation to this compound as it is not a component of its chemical structure.
Stereochemical Requirements in the P3/P4 Region
Stereochemistry in the P3/P4 region of this compound derivatives is crucial for their inhibitory activity. SAR studies revealed that the (S) stereochemical configuration at the stereocenters in the P3/P4 region is essential for potent inhibition. Analogs containing (R)-amino acids at either stereocenter in this region showed significantly reduced potency.
Furthermore, the presence of hydrogen bond donors in the P3/P4 region is vital for maintaining potency. For example, methylation of the cyclohexyl glycine amine amide in this region resulted in a complete loss of inhibitory activity. In contrast, methylation of the terminal amide had no significant effect on potency. Additionally, inhibitors with truncated P3/P4 chains exhibited reduced potency, emphasizing the importance of this region for optimal binding and activity.
Comparative Analysis of Cyclopentane versus Cyclopentene Analogs
During the optimization of this compound, a comparative analysis of cyclopentane and cyclopentene-containing analogs was undertaken. Generally, cyclopentene analogs demonstrated greater potency compared to their cyclopentane counterparts. For instance, a simple norvaline P1 analog with a cyclopentene core (analog 36) was a 100 nM inhibitor against genotype 1a (gt1a) NS3/4A protease, whereas the corresponding cyclopentane analog (analog 29) showed significantly lower potency with a Kᵢ of 2.3 μM.
Despite the observed higher potency of cyclopentene analogs, the decision was made to focus on cyclopentane analogs due to synthetic challenges associated with cyclopentene compounds, including the potential for irreversible covalent attachment via a Michael acceptor in the core.
Table 1: Comparative Potency of Cyclopentane vs. Cyclopentene Analogs against HCV NS3/4A Protease (gt1a)
| Compound Type | Analog | NS3 1a Kᵢ (μM) |
| Cyclopentane | 29 | 2.3 |
| Cyclopentene | 36 | 0.1 |
| Cyclopentene | 37 | 0.0013 |
Table 2: Influence of P3/P4 Stereochemistry and Modifications on NS3 1a Inhibition
| Compound | P3/P4 Stereochemistry/Modification | NS3 1a Kᵢ (μM) / % Inhibition @ 10 μM |
| 29 | (S)-stereocenter | 2.3 |
| 30 | (R)-amino acid at one stereocenter | 35% @ 10 μM |
| 31 | (R)-amino acid at other stereocenter | 37% @ 10 μM |
| 33 | Unmodified P3/P4 | 0.022 |
| 34 | Terminal amide methylation | 0.016 |
| 35 | Cyclohexyl glycine amine amide methylation | >10 |
Computational Approaches to Binding Analysis
Computational approaches, particularly molecular docking and molecular dynamics simulations, have been extensively employed to elucidate the binding mechanisms of this compound with its target proteins. These methods provide atomic-level insights into the drug-target interactions, aiding in the understanding of its efficacy and potential against various viral proteases.
Molecular Docking Simulations of this compound and Analogs
This compound, a macrocyclic, non-covalent, and reversible inhibitor, primarily targets the Hepatitis C Virus (HCV) nonstructural protein 3/4A (NS3/4A) serine protease, binding to its active site and thereby inhibiting viral replication mims.commims.comresearchgate.netdrugbank.com. Molecular docking simulations have been crucial in characterizing these interactions and predicting its binding affinity.
Studies on HCV NS3/4A protease have shown that this compound maintains a consistent binding pattern across different genotypes, such as 1b and 4a. In genotype 1b, this compound interacts with residues including H57, K136, G137, S139, and R155. For genotype 4a, interactions are observed with Q41, H57, K136, G137, and S139. Notably, this compound's ability to bind to the catalytic triad residues H57 and S139 contributes to its activity across these genotypes longdom.org.
Beyond HCV, this compound has been investigated through molecular docking for its potential inhibitory activity against other viral proteases, particularly the SARS-CoV-2 main protease (Mpro) and papain-like protease (PLpro) tandfonline.comujpronline.comujpronline.comnih.govchemrxiv.orgnih.govbiorxiv.orgnih.govacs.orgresearchgate.net. Docking simulations have consistently predicted favorable binding affinities for this compound with SARS-CoV-2 Mpro. Reported binding scores and free energies vary across studies depending on the specific docking algorithm and protein structure used:
| Target Protein | Binding Affinity (kcal/mol) | Interaction Details | Reference |
| SARS-CoV-2 Mpro | -4.686 (Glide score), -81.74 (Binding Free Energy) | Stabilized by 2 hydrogen bonds with Cys 44 and Gly 143; 3 hydrogen bonds with Gly 143, Ser 144, and Cys 145 maintained during MD tandfonline.com | tandfonline.com |
| SARS-CoV-2 Mpro | -6.23 to -7.65 (for this compound and degradation products) | Acidic degradant 2 showed best affinity ujpronline.comujpronline.com | ujpronline.comujpronline.com |
| SARS-CoV-2 Mpro | -11.33 | Three hydrogen bonds with Asn 119, His 163, and Thr 26; three sigma and pi interactions with His 41, Cys 145, and Met 49 nih.gov | nih.gov |
| SARS-CoV-2 3CLpro | -9.4 | Best 3CLpro binding drug in SARS-CoV2 among tested FDA-approved drugs chemrxiv.org | chemrxiv.org |
| SARS-CoV-2 Mpro | -9.9 | Putative binding mode with apo protein crystal structure (PDB ID: 6YB7) nih.govbiorxiv.orgacs.org | nih.govbiorxiv.orgacs.org |
| SARS-CoV-2 3CLpro | -8.78 | Identified as potential inhibitor nih.gov | nih.gov |
The interaction of this compound with SARS-CoV-2 Mpro active site is often characterized by hydrogen bonds and hydrophobic contacts. For instance, hydrogen bonds have been observed with Cys 44 and Gly 143 tandfonline.com. During molecular dynamics simulations, three hydrogen bonds with Gly 143, Ser 144, and Cys 145 of Mpro were consistently maintained tandfonline.com. Other studies reported hydrogen bonds with Asn 119, His 163, and Thr 26, alongside sigma and pi interactions with His 41, Cys 145, and Met 49 nih.gov.
Comparative docking studies have also involved this compound's degradation products against SARS-CoV-2 Mpro, revealing that acidic degradant 2 exhibited superior binding affinity compared to this compound itself and a natural ligand ujpronline.comujpronline.com. Other NS3/4A protease inhibitors like Paritaprevir and Vaniprevir have been compared with this compound in their efficacy against different HCV genotypes, with this compound generally retaining activity across genotypes longdom.orgnih.gov.
Electric Field Calculations for Structure-Function Relationships
Electric field calculations represent a sophisticated computational approach used to establish explicit links between molecular structure and function, particularly in understanding protein-ligand interactions and enzyme catalysis arxiv.org. By projecting electric fields onto specific bonds or atoms within a molecular system, researchers can identify the components that contribute to stabilizing intermolecular interactions, both covalent and non-covalent, within an active site arxiv.org. This method can significantly narrow the exploration space in drug discovery by pinpointing key residues that exert functional consequences on a bound ligand arxiv.org.
While electric field calculations are a valuable tool in computational biophysics for analyzing electrostatics-driven events and understanding how biomolecules create suitable electrostatic environments for biological purposes, specific detailed research findings applying electric field calculations directly to this compound's interaction with HCV NS3/4A protease or other target proteins were not found in the provided search results researchgate.netnih.gov. However, the general methodology suggests that such calculations could provide deeper insights into the precise electrostatic contributions of this compound's chemical moieties to its binding affinity and inhibitory mechanism.
Analysis of Active Site Flexibility
The flexibility of the active site of target proteins plays a crucial role in drug binding and efficacy, and this aspect is often investigated using molecular dynamics (MD) simulations. For this compound, MD simulations have provided insights into its interaction with both HCV NS3/4A protease and SARS-CoV-2 Mpro.
For HCV NS3/4A protease, the flexibility of the active site is known to influence the susceptibility to drug resistance mutations. Inhibitors, including this compound, that feature a P1-P3 macrocycle are more prone to resistance mutations at residue S138, which is located near the active site nih.gov. This highlights the importance of understanding active site flexibility in the development of robust antiviral agents with higher barriers to drug resistance.
Molecular Mechanisms of Antiviral Resistance to Simeprevir
Identification and Characterization of Resistance-Associated Amino Acid Substitutions (RAASs) in HCV NS3 Protease
Both in vitro selection studies and analyses of clinical patient samples have pinpointed several key amino acid substitutions within the NS3 protease that lead to reduced susceptibility to simeprevir. These include mutations at positions F43, Q80, S122, R155, A156, and D168. fda.govfda.gov
The NS3 Q80K polymorphism is a naturally occurring variant with a notable impact on this compound susceptibility, particularly in HCV genotype 1a. brieflands.comasm.orgriberasalud.com Its prevalence exhibits geographical variability, being considerably higher in the United States (approximately 40-50%) compared to Europe (around 19-20%). riberasalud.comdovepress.comoup.com In individuals infected with HCV genotype 1a, the presence of the Q80K polymorphism at baseline has been linked to significantly lower sustained virologic response (SVR) rates when this compound was administered in combination with pegylated-interferon and ribavirin. For instance, SVR rates were reported to be 58% in genotype 1a-infected treatment-naïve patients with Q80K, versus 84% in those without it. asm.org While the Q80K substitution alone typically confers a low-level resistance to this compound, characterized by a fold change (FC) in EC50 between 2 and 50, its presence can lower the resistance barrier, thereby facilitating the emergence of additional resistance mutations, such as R155K, which can lead to higher rates of treatment failure. asm.orgdovepress.comnih.govhivclinic.caeuropa.euresearchgate.net
Table 1: Prevalence and Impact of NS3 Q80K Polymorphism on this compound Susceptibility
| Genotype/Subtype | Prevalence of Q80K (Baseline) | Impact on this compound Susceptibility (Fold Change in EC50) | Clinical Outcome (SVR rates with PEG-IFN/RBV) | Key Observations |
| HCV Genotype 1a | ~30-50% (US), ~19-20% (Europe) riberasalud.comdovepress.comoup.com | Low-level resistance (FC 2-50) nih.govhivclinic.caeuropa.eu | Reduced SVR rates (e.g., 58% vs 84% in GT1a treatment-naïve) asm.org | Facilitates emergence of other mutations (e.g., R155K), leading to higher treatment failure asm.orgdovepress.comresearchgate.net |
| HCV Genotype 1b | ~0.5% dovepress.comdrugbank.com | Generally no significant impact dovepress.com | Not clinically significant due to low prevalence dovepress.com | Rarely observed dovepress.com |
Amino acid substitutions at NS3 positions S122, R155, and D168 are also crucial in conferring resistance to this compound. drugbank.comfda.govrcsb.org
The pattern of emerging resistance mutations often displays genotype-specific profiles. In patients infected with HCV genotype 1a, the R155K substitution is frequently detected, either alone or in conjunction with other mutations at NS3 positions 80 and/or 168. This mutation confers high-level resistance to this compound, with a fold change in EC50 typically greater than 50. dovepress.comnih.govhivclinic.catandfonline.com Specifically, the R155K mutation can lead to a median FC of 88 in a genotype 1a replicon backbone. nih.gov Conversely, for HCV genotype 1b, the D168V mutation is the most commonly observed at the point of treatment failure, also imparting high-level resistance (FC ≥ 50). dovepress.comnih.govhivclinic.catandfonline.com The D168V mutation has been demonstrated to cause a substantial reduction in this compound susceptibility, with FC values ranging from approximately 1,800 to 2,830-fold. nih.govtandfonline.com Other D168 substitutions, such as D168A, D168H, and D168T, similarly confer high-level resistance. nih.gov
Table 2: Genotype-Specific Resistance Profiles for this compound
| Genotype/Subtype | Common Resistance-Associated Substitutions (RAASs) | Level of Resistance (Fold Change in EC50) | Key Observations |
| HCV Genotype 1a | R155K (alone or with Q80/D168) dovepress.comtandfonline.com | High-level (FC ≥ 50, e.g., median 88 for R155K) nih.govhivclinic.ca | Most frequently observed at failure in GT1a dovepress.com |
| HCV Genotype 1b | D168V (most common) dovepress.comtandfonline.com | High-level (FC ≥ 50, e.g., 1,800-2,830 for D168V) nih.govhivclinic.ca | Primarily observed in GT1b isolates dovepress.com |
The D168Q polymorphism is a prevalent natural variant found in nearly all HCV genotype 3a isolates. nih.govagidei.orgnih.gov This naturally occurring substitution confers significant resistance to this compound, with reported reductions in activity exceeding 700-fold in a genotype 1b replicon assay, and up to 700-fold resistance in genotype 3a. nih.govagidei.orgnih.govsemanticscholar.org This inherent resistance largely explains why current NS3 protease inhibitors, including this compound, are primarily effective against genotype 1 and demonstrate reduced efficacy against genotype 3. nih.gov
Amino Acid Substitutions at NS3 Positions S122, R155, and D168
Genotype-Specific Resistance Profiles (e.g., R155K in 1a, D168E/V in 1b)
Cross-Resistance Profiles with Other NS3/4A Protease Inhibitors
Cross-resistance is an expected phenomenon among NS3/4A protease inhibitors due to their common target. fda.gov this compound exhibits cross-resistance with first-generation NS3 protease inhibitors such as telaprevir and boceprevir. dovepress.comhivclinic.ca For example, mutations at amino acid positions 155 and 156 can impact the antiviral activity of this compound, boceprevir, and telaprevir. dovepress.comhivclinic.ca The Q80K polymorphism, while primarily affecting this compound, can also influence the activity of other NS3/4A protease inhibitors like asunaprevir and sovaprevir. agidei.orgnih.gov However, some studies indicate that while this compound may select for D168 mutations, other NS3/4A PIs might not induce the same boosted replication of Q80K+R155K variants, suggesting nuanced differences in their cross-resistance profiles. asm.orgnih.gov
Table 3: Overlapping Mutations Affecting Antiviral Activity of NS3/4A Protease Inhibitors
| NS3 Position | This compound hivclinic.ca | Boceprevir hivclinic.ca | Telaprevir hivclinic.ca |
| V36 | M | A/M | - |
| F43 | C/S | - | - |
| T54 | A | A/S | - |
| R155 | G/I/K/M/Q/T | K/T | - |
| A156 | S/T/V | S/T/V | - |
Molecular Basis of Reduced Viral Susceptibility
The molecular basis for reduced viral susceptibility to this compound involves specific structural alterations within the NS3/4A protease that diminish the inhibitor's binding affinity or efficacy. This compound is a macrocyclic, reversible, and noncovalent inhibitor that binds to the active site of the NS3/4A protease. drugbank.com Resistance mutations frequently emerge at crucial residues located within or in close proximity to this active site. For instance, mutations at D168, a this compound-specific amino acid position, lead to substantial reductions in susceptibility, characterized by high fold changes in EC50. dovepress.comnih.govdrugbank.com The D168V mutation, for example, results in a significant loss of this compound activity, likely due to conformational changes that impede optimal drug binding. nih.govtandfonline.com Similarly, the R155K mutation, particularly in genotype 1a, also confers high-level resistance, underscoring its critical role in the interaction between the protease and this compound. dovepress.comnih.gov The Q80K polymorphism, while conferring lower-level resistance individually, can establish a lower resistance barrier, thereby facilitating the emergence of additional mutations such as R155K, which then contribute to higher rates of treatment failure. asm.orgdovepress.comresearchgate.net These resistance-associated substitutions can alter the precise shape of the protease active site, thereby reducing the drug's ability to bind effectively and allowing the virus to continue its replication cycle. nih.gov
Evolution of Resistance Variants in In Vitro Models
In vitro studies have been instrumental in characterizing the evolution of HCV resistance to this compound. These investigations typically employ replicon-containing human hepatoma cell lines, such as those derived from HCV genotype 1a and 1b, subjected to selective pressure from this compound at either constant or escalating concentrations dovepress.com. Such experimental conditions mimic the selective environment encountered by the virus during drug exposure, leading to the emergence of specific amino acid substitutions within the NS3/4A protease.
Detailed research findings indicate that in a significant majority of cultures (105 out of 109), one or more mutations were observed at key NS3 protease amino acid positions: F43, Q80, R155, A156, and/or D168 . These positions represent critical sites within the NS3/4A protease where alterations can diminish the binding affinity or inhibitory efficacy of this compound.
Key Resistance-Associated Substitutions (RASs) and Their Impact:
D168 Substitutions : Mutations at position D168 were the most frequently observed in vitro, including D168V and D168A, alongside less common substitutions such as E, H, I, T, G, N, or Y . The D168Q polymorphism is also notable, occurring naturally in HCV genotype 3a and leading to a substantial reduction in this compound activity, with a reported >700-fold decrease in susceptibility in a genotype 1b replicon assay dovepress.comasm.org. In wild-type virus, this compound treatment preferentially selected D168A/V mutations asm.orgnih.gov.
Q80K Substitution : The Q80K polymorphism is frequently detected in HCV genotype 1a, with prevalence rates ranging from 19% to 48% mdpi.com. In vitro studies have demonstrated that Q80K confers a low-level resistance, typically reducing this compound susceptibility by approximately 10-fold drugbank.commdpi.com. While its direct impact on in vitro activity may be limited, the presence of Q80K can facilitate the emergence of additional amino acid substitutions, contributing to higher treatment failure rates nih.govnih.gov.
R155K Substitution : In variants harboring the Q80K or Q80R polymorphism, an additional mutation at residue R155, specifically R155K, was selected at this compound concentrations below 2.5 μM asm.orgnih.gov. This suggests a synergistic effect where the initial Q80K/R mutation primes the virus for the acquisition of R155K under drug pressure.
Other Notable Mutations : Less frequent but still significant substitutions observed in vitro include F43S, Q80R, Q80H, A156V, A156T, and A156G . Mutations at S122 and I/V170T have also been identified as reducing susceptibility to this compound drugbank.comnih.gov.
The impact of these resistance-associated substitutions is often quantified by the fold change (FC) in the half-maximal effective concentration (EC50) of this compound. In vitro, clinical isolates without significant resistance mutations generally show full susceptibility (FC ≤ 2.0) or low-level resistance (FC > 2.0 and < 50). However, treatment failure has been consistently associated with the emergence of high-level resistance variants, often exhibiting a median FC value of approximately 400 nih.gov.
Furthermore, some in vitro studies have revealed an unexpected phenomenon: this compound concentrations below 1 μM significantly boosted the replication of Q80K/R R155K variants. Notably, the replication capacity of the Q80K+R155K variant at 0.75 to 1 μM this compound was observed to be higher than that of the wild-type virus in the absence of the drug. This replication boost was specific to this compound and was not observed with other NS3/4A protease inhibitors or in genotype 1b isolates asm.orgnih.gov. This finding provides insight into how certain RAVs can contribute to treatment failure by maintaining or even enhancing viral fitness under drug pressure.
The following table summarizes key resistance-associated substitutions identified in in vitro models and their reported impact on this compound susceptibility:
| NS3 Amino Acid Position | Common Substitutions | Associated Genotypes | In Vitro Impact on this compound Susceptibility |
| D168 | D168V, D168A, D168Q | 1a, 1b, 3a | Most common, high-level resistance. D168Q (natural polymorphism in GT3a) leads to >700-fold reduction dovepress.comasm.orgasm.orgnih.gov. |
| Q80 | Q80K, Q80R, Q80H | 1a | Low-level resistance (approx. 10-fold reduction); facilitates emergence of other mutations drugbank.comasm.orgnih.govmdpi.com. |
| R155 | R155K | 1a | Selected in Q80K/R variants; contributes to resistance asm.orgnih.gov. |
| A156 | A156V, A156T, A156G | 1a, 1b | Observed in resistance selection studies . |
| F43 | F43S | 1a, 1b | Observed in resistance selection studies . |
| S122 | S122R | 1a | Associated with reduced susceptibility drugbank.com. |
| I/V170 | I/V170T | 1a | Associated with reduced susceptibility nih.gov. |
Preclinical Pharmacological Research on Simeprevir
Preclinical Pharmacokinetic Research
Distribution Studies in Non-Human Models (e.g., Liver:Blood Ratio in Rat)
Preclinical studies in animal models have demonstrated that simeprevir undergoes extensive distribution, particularly to the gut and liver tissues. drugbank.comfda.govwikidoc.org In rats, a liver:blood ratio of 29:1 was observed, indicating significant accumulation within the liver. drugbank.comfda.govwikidoc.orgnih.gov While human distribution into compartments other than plasma has not been fully evaluated, preclinical findings in humans showed high tissue/plasma AUC ratios, with the small intestine exhibiting a ratio of 128 and the liver a ratio of 39. nih.gov this compound is largely confined to the plasma rather than the cellular components of the blood, as indicated by a blood:plasma ratio of approximately 0.66. fda.gov
Table 1: this compound Distribution Ratios in Preclinical Models
| Model/Tissue | Ratio (Tissue:Blood/Plasma) | Source |
| Rat Liver | 29:1 (Liver:Blood) | drugbank.comfda.govwikidoc.orgnih.gov |
| Human Liver | 39:1 (Liver:Plasma) | nih.gov |
| Human Small Intestine | 128:1 (Small Intestine:Plasma) | nih.gov |
| Human Blood | ~0.66 (Blood:Plasma) | fda.gov |
Hepatic Uptake Mechanisms: Role of Organic Anion Transporting Polypeptides (OATP1B1/3, OATP2B1)
Hepatic uptake of this compound is primarily mediated by organic anion transporting polypeptides (OATP) 1B1 and OATP1B3. drugbank.comfda.govwikidoc.orgnih.govfda.govnih.gov In vitro data and physiologically-based pharmacokinetic (PBPK) modeling and simulations suggest that this active uptake process is a major mechanism contributing to the distribution of this compound into the liver. fda.govwikidoc.orgfda.gov OATP2B1 has also been identified as a substrate for this compound. fda.gov The saturation of OATP1B1/3-mediated hepatic uptake is a significant factor contributing to the nonlinear pharmacokinetics observed with this compound. fda.govnih.govnih.govresearchgate.net Furthermore, in vitro studies have indicated that this compound can inhibit the uptake transporters OATP1B1, OATP1B3, and OATP2B1. nih.govnih.govfda.govresearchgate.net
Elimination Pathways: Predominance of Biliary Excretion
This compound is predominantly eliminated from the body through biliary excretion. drugbank.comfda.govnih.govnih.govfda.govhivclinic.caeuropa.eudrugs.commims.comwvu.edu Renal clearance plays an insignificant role in its elimination. nih.goveuropa.eu A radioactivity study demonstrated that following a single oral administration of radiolabeled this compound, approximately 91% of the total radioactivity was recovered in the feces, while less than 1% was detected in the urine. drugbank.comnih.govfda.govhivclinic.caeuropa.eudrugs.commims.comwvu.edu
Excretion of Unchanged Drug versus Metabolites
In the feces, the unchanged form of this compound accounted for approximately 31% of the total administered dose. drugbank.comnih.govfda.govhivclinic.caeuropa.eudrugs.commims.comwvu.edu In plasma, unchanged this compound was the primary circulating substance, representing about 85% of the circulating radioactivity 24 hours post-dose. fda.govfda.gov One minor metabolite, M21, was identified in plasma, constituting approximately 8% of the this compound area under the curve (AUC). fda.govhivclinic.ca In feces, M21, in combination with M22, was a major metabolite, accounting for 25.9% of the dose (with an M21/M22 ratio of 60/40). fda.gov These metabolites, M21 and M22, are formed through oxidation on the macrocyclic moiety of this compound. fda.govfda.govhivclinic.caeuropa.eu Several other metabolites were also identified in feces, though none individually comprised more than 6% of the dose. fda.gov No accumulation of metabolites has been observed following multiple-dose administration of this compound. hivclinic.ca
Protein Binding Characteristics in Plasma (>99.9%)
This compound exhibits extensive binding to plasma proteins, with more than 99.9% of the drug bound. drugbank.comfda.govwikidoc.orgnih.govfda.govhivclinic.cadrugs.commims.com The primary plasma protein responsible for this binding is albumin, with alpha 1-acid glycoprotein contributing to a lesser extent. drugbank.comfda.govwikidoc.orgnih.govhivclinic.cadrugs.commims.com This high plasma protein binding is not significantly altered in patients with renal or hepatic impairment. fda.govwikidoc.orgnih.gov
Table 2: this compound Plasma Protein Binding
| Characteristic | Value | Primary Binding Proteins | Source |
| Plasma Protein Binding | >99.9% | Albumin, Alpha 1-acid glycoprotein | drugbank.comfda.govwikidoc.orgnih.govfda.govhivclinic.cadrugs.commims.com |
Nonlinear Pharmacokinetics Attributed to Saturation of Hepatic Uptake and Metabolism
This compound demonstrates nonlinear pharmacokinetics at therapeutic doses. fda.govnih.govnih.govresearchgate.nethivclinic.ca This nonlinearity is primarily attributed to the saturation of both hepatic uptake, mediated by OATP1B1/3, and hepatic metabolism, predominantly by the CYP3A4 enzyme. fda.govnih.govnih.govresearchgate.nethivclinic.ca At higher doses (e.g., above 100 mg once daily in healthy subjects and 75 mg once daily in HCV-infected patients), the maximum plasma concentrations (Cmax) and area under the plasma concentration-time curve (AUC) values increase more than dose-proportionally. nih.govfda.govnih.govresearchgate.net The interplay between hepatic uptake and CYP3A4 metabolism is a key factor in the observed nonlinear pharmacokinetics of this compound. nih.govresearchgate.netdoi.org
Drug Metabolism Research
This compound undergoes hepatic metabolism. drugbank.comfda.govwikidoc.org The primary metabolic pathway involves oxidation mediated by the CYP3A system. drugbank.comfda.govwikidoc.orgnih.govresearchgate.nethivclinic.caeuropa.eudrugs.commims.comeuropa.eu While CYP3A4 plays a major role, the involvement of CYP2C8 and CYP2C19 in this compound metabolism cannot be excluded. drugbank.comfda.govwikidoc.orgfda.goveuropa.eudrugs.commims.comeuropa.eu In animal studies, more than 20 metabolites were identified, but the unchanged drug remains the main component in rat, dog, and human plasma. Metabolites identified in feces, such as M21 and M22, are formed through oxidation on the macrocyclic and/or aromatic moiety, and by O-demethylation followed by oxidation. fda.govhivclinic.caeuropa.eu Despite metabolism, no accumulation of metabolites has been observed after multiple-dose administration of this compound. hivclinic.ca
Q & A
Q. How can in vitro resistance profiles inform clinical trial inclusion criteria?
- Methodological Guidance : Exclude genotype 1a patients with Q80K polymorphisms (prevalence ~30% in US/EU) due to 11-fold reduced this compound susceptibility. For genotype 1b, screen for L31 RAS, which reduces SVR by 50% in non-responders .
Tables for Key Findings
| Parameter | This compound + PR | Sofosbuvir + PR | Telaprevir + PR |
|---|---|---|---|
| SVR in IL28B CC (naïve) | 95% | 98% | 71% |
| SVR in F3–F4 fibrosis | 69% | 85% | 62% |
| Treatment Duration (weeks) | 24 (RGT) | 12 | 48 |
| Resistance Mutation | Fold Change (EC₅₀) | Clinical Impact |
|---|---|---|
| Q80K (1a) | 11x | Avoid this compound use |
| D168V/E (1a/1b) | ≥50x | High-level resistance |
| L31M (1b) | 5x | Moderate relapse risk |
Retrosynthesis Analysis
AI-Powered Synthesis Planning: Our tool employs the Template_relevance Pistachio, Template_relevance Bkms_metabolic, Template_relevance Pistachio_ringbreaker, Template_relevance Reaxys, Template_relevance Reaxys_biocatalysis model, leveraging a vast database of chemical reactions to predict feasible synthetic routes.
One-Step Synthesis Focus: Specifically designed for one-step synthesis, it provides concise and direct routes for your target compounds, streamlining the synthesis process.
Accurate Predictions: Utilizing the extensive PISTACHIO, BKMS_METABOLIC, PISTACHIO_RINGBREAKER, REAXYS, REAXYS_BIOCATALYSIS database, our tool offers high-accuracy predictions, reflecting the latest in chemical research and data.
Strategy Settings
| Precursor scoring | Relevance Heuristic |
|---|---|
| Min. plausibility | 0.01 |
| Model | Template_relevance |
| Template Set | Pistachio/Bkms_metabolic/Pistachio_ringbreaker/Reaxys/Reaxys_biocatalysis |
| Top-N result to add to graph | 6 |
Feasible Synthetic Routes




Featured Recommendations
| Most viewed |


|
|
|---|---|---|
| Most popular with customers |
|
Disclaimer and Information on In-Vitro Research Products
Please be aware that all articles and product information presented on BenchChem are intended solely for informational purposes. The products available for purchase on BenchChem are specifically designed for in-vitro studies, which are conducted outside of living organisms. In-vitro studies, derived from the Latin term "in glass," involve experiments performed in controlled laboratory settings using cells or tissues. It is important to note that these products are not categorized as medicines or drugs, and they have not received approval from the FDA for the prevention, treatment, or cure of any medical condition, ailment, or disease. We must emphasize that any form of bodily introduction of these products into humans or animals is strictly prohibited by law. It is essential to adhere to these guidelines to ensure compliance with legal and ethical standards in research and experimentation.
