Pyrimethamine
Description
Historical Context of Pyrimethamine Discovery and Early Research Applications
The discovery of this compound is a landmark achievement in the history of medicinal chemistry, credited to the pioneering work of Nobel laureates Gertrude B. Elion and George H. Hitchings. sciencehistory.orgnobelprize.org Their research, conducted at Burroughs-Wellcome, was a departure from the traditional trial-and-error method of drug discovery. sciencehistory.org Instead, they championed a "rational drug design" approach, which involved understanding the metabolic pathways of target organisms and creating molecules to specifically interfere with them. sciencehistory.orgacs.org
This strategic approach led them to investigate the synthesis of nucleic acids, essential for the growth and replication of cells. sciencehistory.org By studying the differences in nucleic acid metabolism between pathogenic organisms and human cells, they aimed to develop drugs with selective toxicity. nobelprize.org Their work on purine and pyrimidine analogs culminated in the synthesis of this compound in 1952. wikipedia.orgwikipedia.org
Initially, this compound was developed as a potent antimalarial agent. sciencehistory.orgnobelprize.orgnobelprize.org It functions as a folic acid antagonist by competitively inhibiting the enzyme dihydrofolate reductase (DHFR). wikipedia.orgpatsnap.commedchemexpress.com This enzyme is crucial for the regeneration of tetrahydrofolic acid, a vital cofactor in the synthesis of DNA and RNA. wikipedia.orgpatsnap.com The high affinity of this compound for the plasmodial DHFR compared to the human enzyme provided the basis for its selective antiprotozoal activity. nobelprize.orgpatsnap.com Early research applications were, therefore, primarily focused on its efficacy against Plasmodium species, the parasites responsible for malaria. wikipedia.orgpatsnap.comguidetopharmacology.org Beyond malaria, its antiprotozoal activity was also explored against other parasites like Toxoplasma gondii, the causative agent of toxoplasmosis. patsnap.commedscape.com
| Key Milestone | Year | Researchers | Significance |
| Synthesis of this compound | 1952 | Gertrude B. Elion & George H. Hitchings | A result of the "rational drug design" approach, targeting nucleic acid synthesis. wikipedia.orgwikipedia.org |
| Medical Use | 1953 | Introduced primarily for the treatment of malaria. wikipedia.org | |
| Nobel Prize in Physiology or Medicine | 1988 | Gertrude B. Elion & George H. Hitchings | Awarded for their discoveries of "important principles for drug treatment," including the work that led to this compound. nobelprize.org |
Evolution of Research Perspectives on this compound: From Antiprotozoal to Broader Biological Activities
While this compound's role as an antiprotozoal agent is well-established, the last few decades have witnessed a significant evolution in research perspectives, unveiling its potential in other therapeutic areas. This shift has been largely driven by the practice of drug repurposing, where existing drugs are investigated for new medical applications. nih.govoup.com
A major area of this expanded research is in oncology . nih.govoup.comnih.gov The rationale for exploring this compound as an anticancer agent stems from its known mechanism of inhibiting dihydrofolate reductase (DHFR), an enzyme that is also vital for the proliferation of cancer cells. nih.govaacrjournals.org Research has shown that this compound can inhibit human DHFR, although its binding affinity is weaker than that of classical chemotherapy drugs like methotrexate. aacrjournals.org
Beyond DHFR inhibition, studies have revealed that this compound's anticancer effects may be mediated through multiple pathways. nih.govresearchgate.net One of the most studied alternative mechanisms is the inhibition of the Signal Transducer and Activator of Transcription 3 (STAT3) signaling pathway. nih.govoup.com The STAT3 protein is often overactive in many cancers, promoting tumor growth, invasion, and metastasis. nih.govoup.com this compound has been shown to inhibit STAT3 function, leading to reduced proliferation and invasion of cancer cells in preclinical models of breast cancer. nih.govresearchgate.net Furthermore, research has demonstrated its ability to induce apoptosis (programmed cell death) in cancer cells through various mechanisms, including those dependent on cathepsin B and caspases. aacrjournals.orgresearchgate.net Other investigated targets and pathways in cancer research include NF-κB, AIMP2-DX2, MAPK, and telomerase. nih.govresearchgate.netmdpi.com
The antiviral potential of this compound has also been a subject of investigation. For instance, in vitro studies have explored its effect on the replication of the rabies virus, where it was found to inhibit the virus, although this effect was not replicated in animal models. pasteur.frresearchgate.net Another study investigated its impact on HIV-1 replication, revealing a complex interaction where it enhanced viral replication in certain cell lines. nih.gov
More recently, research has explored the potential of this compound in treating genetic disorders. In 2011, researchers discovered that this compound can increase the activity of the β-hexosaminidase A enzyme, which is deficient in late-onset Tay-Sachs disease. wikipedia.orgclinicaltrials.govclinicaltrials.gov This has led to clinical investigations into its potential to slow the progression of this neurodegenerative disorder. clinicaltrials.govclinicaltrials.gov It is also being evaluated for its potential in treating amyotrophic lateral sclerosis (ALS) due to its ability to reduce the expression of SOD1, a key protein implicated in some forms of the disease. wikipedia.org
Significance of this compound in Current Biomedical Research Paradigms
This compound continues to be a significant molecule in contemporary biomedical research, largely due to the ongoing efforts in drug repurposing. mdpi.com Its well-established safety profile from decades of clinical use as an antiprotozoal agent makes it an attractive candidate for investigation in new therapeutic contexts. nih.gov
In oncology research , this compound is being actively studied as a potential anticancer agent, both as a standalone therapy and in combination with other drugs. nih.govmdpi.comnih.gov Systematic reviews of preclinical studies have highlighted its ability to reduce tumor burden in various cancer models. nih.govoup.com Current research is focused on elucidating its precise mechanisms of action in different cancer types and identifying biomarkers that could predict patient response. researchgate.netmdpi.com For example, studies are investigating its efficacy in cancers with high expression of specific proteins like AIMP2-DX2 or those dependent on the STAT3 signaling pathway. mdpi.com The potential for synergistic effects when combined with existing chemotherapy agents like temozolomide for glioblastoma is also an active area of research. ugent.beugent.be
In the field of infectious diseases , while resistance has limited its use for malaria in many regions, research continues on its activity against other pathogens. malariaworld.orgfrontiersin.orgplos.org For instance, recent in vitro studies have shown that this compound exhibits superior activity against Gardnerella species, a key bacterium associated with bacterial vaginosis, when compared to standard treatments. oup.comnih.gov This suggests a potential new application for this compound in women's health. oup.comnih.gov Furthermore, its role in combination therapies for toxoplasmosis remains a standard of care, and research into optimizing these regimens continues. patsnap.commdpi.com
The exploration of this compound in rare diseases represents a promising frontier. Clinical trials are underway to evaluate its efficacy in slowing the progression of GM2 gangliosidoses, such as Tay-Sachs and Sandhoff disease, based on its ability to act as a pharmacological chaperone for the deficient enzyme. clinicaltrials.govclinicaltrials.gov Its potential role in treating amyotrophic lateral sclerosis (ALS) is also under investigation. wikipedia.org
The table below summarizes some of the current research applications of this compound being investigated.
| Research Area | Investigated Application | Key Findings/Rationale |
| Oncology | Repurposing as an anticancer agent for various cancers (e.g., lung, breast, leukemia, glioblastoma). nih.govnih.govmdpi.comugent.be | Inhibition of DHFR, STAT3, and other oncogenic pathways; induction of apoptosis. nih.govnih.govaacrjournals.orgmdpi.com |
| Infectious Diseases | Alternative treatment for bacterial vaginosis. oup.comnih.gov | Superior in vitro activity against Gardnerella species compared to standard antibiotics. oup.comnih.gov |
| Combination therapy for toxoplasmosis. patsnap.commdpi.com | Synergistic effect with sulfonamides in inhibiting the folic acid pathway of Toxoplasma gondii. patsnap.com | |
| Rare Genetic Diseases | Treatment for late-onset Tay-Sachs disease and Sandhoff disease (GM2 gangliosidoses). clinicaltrials.govclinicaltrials.gov | Acts as a pharmacological chaperone to increase the activity of the deficient β-hexosaminidase A enzyme. clinicaltrials.govclinicaltrials.gov |
| Potential treatment for amyotrophic lateral sclerosis (ALS). wikipedia.org | Reduces the expression of the SOD1 protein. wikipedia.org |
Properties
IUPAC Name |
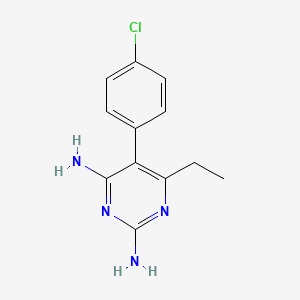
5-(4-chlorophenyl)-6-ethylpyrimidine-2,4-diamine |
Source


|
|---|---|---|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
InChI |
InChI=1S/C12H13ClN4/c1-2-9-10(11(14)17-12(15)16-9)7-3-5-8(13)6-4-7/h3-6H,2H2,1H3,(H4,14,15,16,17) |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
InChI Key |
WKSAUQYGYAYLPV-UHFFFAOYSA-N |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Canonical SMILES |
CCC1=C(C(=NC(=N1)N)N)C2=CC=C(C=C2)Cl |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Molecular Formula |
C12H13ClN4 |
Source
|
| Record name | PYRIMETHAMINE | |
| Source | CAMEO Chemicals | |
| URL | https://cameochemicals.noaa.gov/chemical/20974 | |
| Description | CAMEO Chemicals is a chemical database designed for people who are involved in hazardous material incident response and planning. CAMEO Chemicals contains a library with thousands of datasheets containing response-related information and recommendations for hazardous materials that are commonly transported, used, or stored in the United States. CAMEO Chemicals was developed by the National Oceanic and Atmospheric Administration's Office of Response and Restoration in partnership with the Environmental Protection Agency's Office of Emergency Management. | |
| Explanation | CAMEO Chemicals and all other CAMEO products are available at no charge to those organizations and individuals (recipients) responsible for the safe handling of chemicals. However, some of the chemical data itself is subject to the copyright restrictions of the companies or organizations that provided the data. | |
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
DSSTOX Substance ID |
DTXSID9021217 |
Source
|
| Record name | Pyrimethamine | |
| Source | EPA DSSTox | |
| URL | https://comptox.epa.gov/dashboard/DTXSID9021217 | |
| Description | DSSTox provides a high quality public chemistry resource for supporting improved predictive toxicology. | |
Molecular Weight |
248.71 g/mol |
Source
|
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Physical Description |
Pyrimethamine is an odorless white crystalline powder. Tasteless. An antimalarial drug., Solid |
Source
|
| Record name | PYRIMETHAMINE | |
| Source | CAMEO Chemicals | |
| URL | https://cameochemicals.noaa.gov/chemical/20974 | |
| Description | CAMEO Chemicals is a chemical database designed for people who are involved in hazardous material incident response and planning. CAMEO Chemicals contains a library with thousands of datasheets containing response-related information and recommendations for hazardous materials that are commonly transported, used, or stored in the United States. CAMEO Chemicals was developed by the National Oceanic and Atmospheric Administration's Office of Response and Restoration in partnership with the Environmental Protection Agency's Office of Emergency Management. | |
| Explanation | CAMEO Chemicals and all other CAMEO products are available at no charge to those organizations and individuals (recipients) responsible for the safe handling of chemicals. However, some of the chemical data itself is subject to the copyright restrictions of the companies or organizations that provided the data. | |
| Record name | Pyrimethamine | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014350 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
Solubility |
>37.3 [ug/mL] (The mean of the results at pH 7.4), less than 0.1 mg/mL at 70 °F (NTP, 1992), In water, 10 mg/L (temperature not specified), Practically insoluble in water; slightly soluble in ethanol (about 9 g/L), in dilute HCl (about 5 g/L); soluble in boiling ethanol (about 25 g/L); Very sparingly soluble in propylene glycol and dimethylacetamide at 70 °C, 1.79e-01 g/L |
Source
|
| Record name | SID56422411 | |
| Source | Burnham Center for Chemical Genomics | |
| URL | https://pubchem.ncbi.nlm.nih.gov/bioassay/1996#section=Data-Table | |
| Description | Aqueous solubility in buffer at pH 7.4 | |
| Record name | PYRIMETHAMINE | |
| Source | CAMEO Chemicals | |
| URL | https://cameochemicals.noaa.gov/chemical/20974 | |
| Description | CAMEO Chemicals is a chemical database designed for people who are involved in hazardous material incident response and planning. CAMEO Chemicals contains a library with thousands of datasheets containing response-related information and recommendations for hazardous materials that are commonly transported, used, or stored in the United States. CAMEO Chemicals was developed by the National Oceanic and Atmospheric Administration's Office of Response and Restoration in partnership with the Environmental Protection Agency's Office of Emergency Management. | |
| Explanation | CAMEO Chemicals and all other CAMEO products are available at no charge to those organizations and individuals (recipients) responsible for the safe handling of chemicals. However, some of the chemical data itself is subject to the copyright restrictions of the companies or organizations that provided the data. | |
| Record name | Pyrimethamine | |
| Source | DrugBank | |
| URL | https://www.drugbank.ca/drugs/DB00205 | |
| Description | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. | |
| Explanation | Creative Common's Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/legalcode) | |
| Record name | Pyrimethamine | |
| Source | Hazardous Substances Data Bank (HSDB) | |
| URL | https://pubchem.ncbi.nlm.nih.gov/source/hsdb/8042 | |
| Description | The Hazardous Substances Data Bank (HSDB) is a toxicology database that focuses on the toxicology of potentially hazardous chemicals. It provides information on human exposure, industrial hygiene, emergency handling procedures, environmental fate, regulatory requirements, nanomaterials, and related areas. The information in HSDB has been assessed by a Scientific Review Panel. | |
| Record name | Pyrimethamine | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014350 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
Color/Form |
Crystals, White scored tablets contains 25 mg pyrimethamine /Daraprim/ | |
CAS No. |
58-14-0 |
Source
|
| Record name | PYRIMETHAMINE | |
| Source | CAMEO Chemicals | |
| URL | https://cameochemicals.noaa.gov/chemical/20974 | |
| Description | CAMEO Chemicals is a chemical database designed for people who are involved in hazardous material incident response and planning. CAMEO Chemicals contains a library with thousands of datasheets containing response-related information and recommendations for hazardous materials that are commonly transported, used, or stored in the United States. CAMEO Chemicals was developed by the National Oceanic and Atmospheric Administration's Office of Response and Restoration in partnership with the Environmental Protection Agency's Office of Emergency Management. | |
| Explanation | CAMEO Chemicals and all other CAMEO products are available at no charge to those organizations and individuals (recipients) responsible for the safe handling of chemicals. However, some of the chemical data itself is subject to the copyright restrictions of the companies or organizations that provided the data. | |
| Record name | Pyrimethamine | |
| Source | CAS Common Chemistry | |
| URL | https://commonchemistry.cas.org/detail?cas_rn=58-14-0 | |
| Description | CAS Common Chemistry is an open community resource for accessing chemical information. Nearly 500,000 chemical substances from CAS REGISTRY cover areas of community interest, including common and frequently regulated chemicals, and those relevant to high school and undergraduate chemistry classes. This chemical information, curated by our expert scientists, is provided in alignment with our mission as a division of the American Chemical Society. | |
| Explanation | The data from CAS Common Chemistry is provided under a CC-BY-NC 4.0 license, unless otherwise stated. | |
| Record name | Pyrimethamine [USP:INN:BAN:JAN] | |
| Source | ChemIDplus | |
| URL | https://pubchem.ncbi.nlm.nih.gov/substance/?source=chemidplus&sourceid=0000058140 | |
| Description | ChemIDplus is a free, web search system that provides access to the structure and nomenclature authority files used for the identification of chemical substances cited in National Library of Medicine (NLM) databases, including the TOXNET system. | |
| Record name | Pyrimethamine | |
| Source | DrugBank | |
| URL | https://www.drugbank.ca/drugs/DB00205 | |
| Description | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. | |
| Explanation | Creative Common's Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/legalcode) | |
| Record name | pyrimethamine | |
| Source | DTP/NCI | |
| URL | https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&outputformat=html&searchlist=757306 | |
| Description | The NCI Development Therapeutics Program (DTP) provides services and resources to the academic and private-sector research communities worldwide to facilitate the discovery and development of new cancer therapeutic agents. | |
| Explanation | Unless otherwise indicated, all text within NCI products is free of copyright and may be reused without our permission. Credit the National Cancer Institute as the source. | |
| Record name | pyrimethamine | |
| Source | DTP/NCI | |
| URL | https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&outputformat=html&searchlist=3061 | |
| Description | The NCI Development Therapeutics Program (DTP) provides services and resources to the academic and private-sector research communities worldwide to facilitate the discovery and development of new cancer therapeutic agents. | |
| Explanation | Unless otherwise indicated, all text within NCI products is free of copyright and may be reused without our permission. Credit the National Cancer Institute as the source. | |
| Record name | Pyrimethamine | |
| Source | EPA DSSTox | |
| URL | https://comptox.epa.gov/dashboard/DTXSID9021217 | |
| Description | DSSTox provides a high quality public chemistry resource for supporting improved predictive toxicology. | |
| Record name | Pyrimethamine | |
| Source | European Chemicals Agency (ECHA) | |
| URL | https://echa.europa.eu/substance-information/-/substanceinfo/100.000.331 | |
| Description | The European Chemicals Agency (ECHA) is an agency of the European Union which is the driving force among regulatory authorities in implementing the EU's groundbreaking chemicals legislation for the benefit of human health and the environment as well as for innovation and competitiveness. | |
| Explanation | Use of the information, documents and data from the ECHA website is subject to the terms and conditions of this Legal Notice, and subject to other binding limitations provided for under applicable law, the information, documents and data made available on the ECHA website may be reproduced, distributed and/or used, totally or in part, for non-commercial purposes provided that ECHA is acknowledged as the source: "Source: European Chemicals Agency, http://echa.europa.eu/". Such acknowledgement must be included in each copy of the material. ECHA permits and encourages organisations and individuals to create links to the ECHA website under the following cumulative conditions: Links can only be made to webpages that provide a link to the Legal Notice page. | |
| Record name | PYRIMETHAMINE | |
| Source | FDA Global Substance Registration System (GSRS) | |
| URL | https://gsrs.ncats.nih.gov/ginas/app/beta/substances/Z3614QOX8W | |
| Description | The FDA Global Substance Registration System (GSRS) enables the efficient and accurate exchange of information on what substances are in regulated products. Instead of relying on names, which vary across regulatory domains, countries, and regions, the GSRS knowledge base makes it possible for substances to be defined by standardized, scientific descriptions. | |
| Explanation | Unless otherwise noted, the contents of the FDA website (www.fda.gov), both text and graphics, are not copyrighted. They are in the public domain and may be republished, reprinted and otherwise used freely by anyone without the need to obtain permission from FDA. Credit to the U.S. Food and Drug Administration as the source is appreciated but not required. | |
| Record name | Pyrimethamine | |
| Source | Hazardous Substances Data Bank (HSDB) | |
| URL | https://pubchem.ncbi.nlm.nih.gov/source/hsdb/8042 | |
| Description | The Hazardous Substances Data Bank (HSDB) is a toxicology database that focuses on the toxicology of potentially hazardous chemicals. It provides information on human exposure, industrial hygiene, emergency handling procedures, environmental fate, regulatory requirements, nanomaterials, and related areas. The information in HSDB has been assessed by a Scientific Review Panel. | |
| Record name | Pyrimethamine | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014350 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
Melting Point |
451 to 453 °F (capillary) (NTP, 1992), 233-234 °C (capillary); 240-242 °C (copper block), 233.5 °C |
Source
|
| Record name | PYRIMETHAMINE | |
| Source | CAMEO Chemicals | |
| URL | https://cameochemicals.noaa.gov/chemical/20974 | |
| Description | CAMEO Chemicals is a chemical database designed for people who are involved in hazardous material incident response and planning. CAMEO Chemicals contains a library with thousands of datasheets containing response-related information and recommendations for hazardous materials that are commonly transported, used, or stored in the United States. CAMEO Chemicals was developed by the National Oceanic and Atmospheric Administration's Office of Response and Restoration in partnership with the Environmental Protection Agency's Office of Emergency Management. | |
| Explanation | CAMEO Chemicals and all other CAMEO products are available at no charge to those organizations and individuals (recipients) responsible for the safe handling of chemicals. However, some of the chemical data itself is subject to the copyright restrictions of the companies or organizations that provided the data. | |
| Record name | Pyrimethamine | |
| Source | DrugBank | |
| URL | https://www.drugbank.ca/drugs/DB00205 | |
| Description | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. | |
| Explanation | Creative Common's Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/legalcode) | |
| Record name | Pyrimethamine | |
| Source | Hazardous Substances Data Bank (HSDB) | |
| URL | https://pubchem.ncbi.nlm.nih.gov/source/hsdb/8042 | |
| Description | The Hazardous Substances Data Bank (HSDB) is a toxicology database that focuses on the toxicology of potentially hazardous chemicals. It provides information on human exposure, industrial hygiene, emergency handling procedures, environmental fate, regulatory requirements, nanomaterials, and related areas. The information in HSDB has been assessed by a Scientific Review Panel. | |
| Record name | Pyrimethamine | |
| Source | Human Metabolome Database (HMDB) | |
| URL | http://www.hmdb.ca/metabolites/HMDB0014350 | |
| Description | The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. | |
| Explanation | HMDB is offered to the public as a freely available resource. Use and re-distribution of the data, in whole or in part, for commercial purposes requires explicit permission of the authors and explicit acknowledgment of the source material (HMDB) and the original publication (see the HMDB citing page). We ask that users who download significant portions of the database cite the HMDB paper in any resulting publications. | |
Molecular and Biochemical Mechanisms of Action
Dihydrofolate Reductase (DHFR) Inhibition
Pyrimethamine's primary mechanism of action is the competitive inhibition of dihydrofolate reductase (DHFR), an essential enzyme in the folate metabolic pathway. targetmol.comwikipedia.orgpatsnap.comnih.gov This inhibition disrupts the synthesis of vital cellular components, ultimately hindering cell proliferation. patsnap.comdrugbank.com
Detailed Mechanism of Competitive Inhibition of DHFR by this compound
This compound acts as a competitive inhibitor of DHFR, meaning it competes with the enzyme's natural substrate, dihydrofolate (DHF), for binding to the active site. patsnap.comasm.org The high affinity of this compound for the DHFR active site allows it to effectively block the binding of DHF. patsnap.com This inhibitory action is highly selective for the plasmodial and Toxoplasma gondii forms of the enzyme over the human counterpart. drugbank.com The binding of this compound to DHFR is a reversible process. nih.gov
Impact on Tetrahydrofolic Acid Regeneration and Downstream Biosynthetic Pathways
The inhibition of DHFR by this compound directly halts the reduction of dihydrofolate (DHF) to tetrahydrofolic acid (THF). wikipedia.orgpatsnap.com THF and its derivatives are crucial one-carbon donors for the synthesis of essential biomolecules. patsnap.com Specifically, the lack of THF disrupts the de novo synthesis of purines and the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), a necessary step in pyrimidine synthesis. nih.govlibretexts.org The ultimate consequence of this disruption is the impaired synthesis of DNA, RNA, and proteins, which are all vital for cell division and growth. patsnap.comgoogle.com
Structural Biology of this compound-DHFR Interactions
The selective toxicity of this compound against parasites like Plasmodium falciparum compared to humans is rooted in the structural differences between their respective DHFR enzymes. aacrjournals.org
In Plasmodium species, this compound binds effectively to the active site of DHFR (pDHFR). The binding is characterized by the pyrimidine ring of the inhibitor being buried within a deep cleft of the enzyme. nih.gov This interaction is stabilized by a combination of hydrogen bonds and van der Waals forces. nih.gov However, mutations in the dhfr gene, particularly at codons 51, 59, 108, and 164, can lead to resistance by introducing steric hindrance and altering the active site's conformation, thereby reducing the binding affinity of this compound. nih.govmdpi.comcambridge.org For instance, the S108N mutation, where serine is replaced by asparagine at position 108, creates steric clashes with the rigid p-chlorophenyl group of this compound, significantly weakening its binding. cambridge.orgproteopedia.org
The binding of this compound within the DHFR active site is a delicate interplay of molecular interactions. Hydrogen bonds are crucial for the initial anchoring of the inhibitor. In Plasmodium vivax DHFR, the N1 and 2-amino nitrogen atoms of this compound's pyrimidine ring form hydrogen bonds with the carboxylate oxygen atoms of Asp-53. nih.gov The 4-amino nitrogen also forms a hydrogen bond with the main chain carbonyl oxygen of Ile-14. nih.gov
Steric hindrance plays a pivotal role in drug resistance. Mutations that introduce bulkier amino acid side chains into the active site can physically obstruct the binding of this compound. mdpi.com The S108N mutation in P. falciparum DHFR is a classic example, where the larger asparagine residue clashes with the p-chlorophenyl group of the drug. cambridge.org This steric conflict can lead to a dislocation of the nicotinamide ring of the NADPH cofactor, further compromising the inhibitor's binding. cambridge.org The loss of hydrogen bonds and the introduction of steric clashes are the primary mechanisms by which mutations in the DHFR gene confer resistance to this compound. wikipedia.org
Interactive Data Table: Key Amino Acid Interactions in this compound Binding
| Enzyme | Key Residue(s) | Type of Interaction | Impact on Binding |
| P. vivax DHFR (Wild-Type) | Asp-53 | Hydrogen Bonding | Stabilizes inhibitor binding nih.gov |
| P. vivax DHFR (Wild-Type) | Ile-14 | Hydrogen Bonding | Stabilizes inhibitor binding nih.gov |
| P. falciparum DHFR (Mutant) | Asn-108 | Steric Hindrance | Reduces binding affinity, leads to resistance cambridge.orgproteopedia.org |
| Human DHFR | Phe31, Gln35, Asn64 | - | Lower binding affinity compared to pDHFR proteopedia.org |
Interactive Data Table: Impact of DHFR Inhibition on Cellular Processes
| Process | Key Molecule(s) | Effect of this compound | Consequence |
| Folate Metabolism | Dihydrofolate (DHF), Tetrahydrofolic Acid (THF) | Inhibition of DHF to THF conversion wikipedia.orgpatsnap.com | Depletion of THF pool pnas.org |
| Purine Synthesis | Purine nucleotides | Disruption of de novo synthesis nih.gov | Impaired DNA and RNA synthesis patsnap.com |
| Pyrimidine Synthesis | Deoxythymidine monophosphate (dTMP) | Inhibition of dTMP synthesis nih.govlibretexts.org | Impaired DNA synthesis patsnap.com |
| Cellular Proliferation | DNA, RNA, Proteins | Inhibition of synthesis patsnap.comgoogle.com | Arrest of cell division and growth patsnap.com |
Comparative Analysis of this compound Binding to Human DHFR (hDHFR)
Alternative Molecular Targets and Pathways
While the primary mechanism of action of this compound involves the inhibition of dihydrofolate reductase (DHFR), extensive research has revealed its engagement with a variety of other molecular targets and signaling pathways. These alternative mechanisms contribute to its broader pharmacological effects, particularly in the context of anticancer activity. This section details these alternative molecular interactions and their downstream consequences.
Thymidine Phosphorylase (TP) Inhibition and Dual Targeting Mechanisms
Recent studies have identified Thymidine Phosphorylase (TP) as a direct target of this compound, unveiling a dual-targeting mechanism that contributes to its antitumor effects. aacrjournals.org While both this compound and the classic DHFR inhibitor, Methotrexate, can suppress cancer cell proliferation by targeting human dihydrofolate reductase (hDHFR), only this compound demonstrates the ability to inhibit epithelial-mesenchymal transition (EMT), metastasis, and invasion of lung cancer cells. aacrjournals.orgnih.gov This distinction suggested that DHFR is not the sole target of this compound's anticancer activity. aacrjournals.org
Further investigation revealed that this compound also targets TP, an enzyme implicated in tumor migration and invasion. aacrjournals.org The structural similarity between this compound and thymidine supports the hypothesis that TP is another target of this drug. aacrjournals.org Molecular docking simulations have shown favorable binding scores for this compound with TP. aacrjournals.org This dual inhibition of both DHFR and TP by this compound plays a synergistic role in its antitumor proliferation and anti-metastatic activities. aacrjournals.orgnih.gov Notably, the overexpression of TP has been associated with a poor prognosis in lung cancer patients, highlighting the clinical relevance of this target. aacrjournals.orgnih.gov
Table 1: Comparative Effects of this compound and Methotrexate on Cancer Cell Processes
| Cellular Process | This compound | Methotrexate | Reference |
|---|---|---|---|
| Proliferation Inhibition | Yes | Yes | aacrjournals.org, nih.gov |
| EMT Inhibition | Yes | No | aacrjournals.org, nih.gov |
| Metastasis/Invasion Inhibition | Yes | No | aacrjournals.org, nih.gov |
| Primary Target(s) | DHFR, TP | DHFR | aacrjournals.org, nih.gov |
Inhibition of Signal Transducer and Activator of Transcription 3 (STAT3) Pathway
This compound has been identified as a novel and specific inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3) transcriptional activity. nih.govnih.govresearchgate.net STAT3 is a key transcription factor that, when persistently activated, promotes many tumorigenic processes. nih.govnih.govoup.com Interestingly, this compound's inhibitory effect on the STAT3 pathway is not direct. It does not significantly affect the primary steps of STAT3 activation, such as STAT3 phosphorylation, its translocation to the nucleus, or its binding to DNA, at concentrations sufficient to block its transcriptional activity. nih.govnih.govresearchgate.net
The mechanism underlying STAT3 inhibition by this compound is a consequence of its primary action on DHFR. nih.govnih.gov By inhibiting DHFR, this compound leads to a deficiency in reduced folates, which are crucial for the synthesis of purines and pyrimidines. nih.govnih.gov This disruption of folate metabolism, specifically the reduction in the intracellular pool of 5,10-methylene-THF needed for thymidylate synthesis, ultimately results in decreased STAT3 transcriptional activity. nih.gov This finding reveals an unexpected link between folate metabolism and the regulation of the STAT3 signaling pathway. nih.govnih.gov The combination of this compound with other STAT3 inhibitors or therapies targeting downstream effectors may offer novel strategies for treating STAT3-driven cancers. nih.govoup.com
Induction of Cathepsin B-Dependent and Caspase-Dependent Apoptotic Pathways
This compound has been shown to induce apoptosis in various cancer cells, including melanoma and pituitary adenoma cells, through a multifaceted mechanism that involves both cathepsin and caspase cascades. aacrjournals.orgmedicinacomplementar.com.brnih.govresearchgate.netaacrjournals.org This dual-edged mechanism allows this compound to bypass or overcome the resistance to apoptosis often observed in cancer cells. nih.gov
In melanoma cells, this compound treatment leads to the activation of both initiator caspases (caspase-8 and caspase-9) and the executioner caspase-3. medicinacomplementar.com.brnih.govnih.gov The activation of caspase-9 suggests the involvement of the intrinsic, mitochondria-mediated apoptotic pathway. medicinacomplementar.com.br Concurrently, this compound induces an early and significant increase in the expression and activity of cathepsin B. medicinacomplementar.com.br The use of specific inhibitors has confirmed the functional roles of both pathways. A pan-caspase inhibitor (Z-VAD-FMK) and a cathepsin B inhibitor (CA-074-Me) can both individually reduce this compound-induced apoptosis, with a more pronounced protective effect observed when both inhibitors are used simultaneously. medicinacomplementar.com.brnih.gov This indicates that both caspase-dependent and cathepsin B-dependent pathways are actively involved in the execution of apoptosis following this compound treatment. medicinacomplementar.com.brnih.gov
Table 2: Key Mediators in this compound-Induced Apoptosis
| Apoptotic Mediator | Effect of this compound | Cell Type(s) | Reference |
|---|---|---|---|
| Caspase-8 | Activation | Melanoma | nih.gov, medicinacomplementar.com.br, nih.gov |
| Caspase-9 | Activation | Melanoma, NSCLC | nih.gov, medicinacomplementar.com.br, nih.gov, iiarjournals.org |
| Caspase-3 | Activation | Melanoma, NSCLC | nih.gov, nih.gov, iiarjournals.org |
| Cathepsin B | Increased Expression/Activity | Melanoma, Pituitary Adenoma | nih.gov, medicinacomplementar.com.br, nih.gov |
| Bcl-2 | Decreased Expression | Prostate Cancer, Pituitary Adenoma | , nih.gov |
| Bax | Upregulated Expression | Pituitary Adenoma | nih.gov |
Modulation of p38-NF-κB Axis
In the context of castration-resistant prostate cancer (CRPC), this compound has been shown to exert its antitumor effects by inhibiting the p38-NF-κB signaling axis. researchgate.netnih.gov The p38 mitogen-activated protein kinase (MAPK) and the nuclear factor-κB (NF-κB) pathways are crucial in cancer development and progression, regulating processes like proliferation, inflammation, and apoptosis. nih.gov
Research has demonstrated that this compound significantly decreases the phosphorylation of both p38 and the p65 subunit of NF-κB in a dose-dependent manner in CRPC cells, without altering the total protein levels of p38. researchgate.net This suggests that this compound acts as an inhibitor of p38 MAPK activity. nih.gov The inhibition of p38 by this compound subsequently impairs the activity of p65/NF-κB. This was further supported by experiments where both this compound and a specific p38 MAPK inhibitor (SB202190) could prevent both basal and TNF-α-induced NF-κB activity. nih.gov Therefore, this compound negatively regulates p65/NF-κB activity in CRPC cells through a mechanism dependent on the inhibition of p38 MAPK. nih.gov
Regulation of Cell Cycle Progression (e.g., G0/G1 and S-phase arrest)
This compound has been demonstrated to inhibit cancer cell proliferation by inducing cell cycle arrest at different phases, depending on the cancer type. nih.govnih.goviiarjournals.orgnih.gov In human metastatic melanoma cells and castration-resistant prostate cancer cells, this compound treatment leads to a significant accumulation of cells in the S-phase of the cell cycle. nih.govnih.govnih.gov This S-phase arrest is a notable mechanism contributing to its antiproliferative effects in these cancers. nih.govnih.gov
Conversely, in non-small cell lung cancer (NSCLC) A549 cells, this compound induces cell cycle arrest at the G0/G1 phase. iiarjournals.org This G0/G1 arrest is associated with the downregulation of key cell cycle regulatory proteins, including cyclin D1, cyclin E, cyclin-dependent kinase 2 (CDK2), and CDK4, along with the upregulation of the CDK inhibitor p21. iiarjournals.org In JAR trophoblastic cells, treatment with this compound also resulted in G0/G1 and S phase arrest. nih.gov The ability of this compound to halt cell cycle progression at these critical checkpoints is a key component of its anticancer activity. iiarjournals.orgnih.gov
Table 3: Cell Cycle Effects of this compound in Different Cancer Cell Lines
| Cancer Type | Phase of Cell Cycle Arrest | Key Molecular Changes | Reference |
|---|---|---|---|
| Metastatic Melanoma | S-phase | Not specified | nih.gov, nih.gov |
| Prostate Cancer (CRPC) | S-phase | Not specified | , nih.gov |
| Non-Small Cell Lung Cancer (A549) | G0/G1 phase | ↓ Cyclin D1, ↓ Cyclin E, ↓ CDK2, ↓ CDK4, ↑ p21 | iiarjournals.org |
| JAR Trophoblastic Cells | G0/G1 and S phase | Not specified | nih.gov |
Induction of Ubiquitination-Mediated Degradation of AIMP2-DX2
A novel anticancer mechanism of this compound involves the targeted degradation of AIMP2-DX2, a splice variant of the aminoacyl-tRNA synthetase-interacting multifunctional protein 2 (AIMP2). frontiersin.orgmdpi.comnih.gov While AIMP2 acts as a tumor suppressor, AIMP2-DX2 is a tumorigenic factor that is frequently overexpressed in lung cancer, and its ratio to AIMP2 correlates with cancer progression. frontiersin.orgmdpi.comnih.gov
This compound has been shown to dose-dependently decrease the protein level of AIMP2-DX2 without affecting the level of the tumor-suppressing AIMP2. mdpi.com Further investigation revealed that this compound promotes the ubiquitination of AIMP2-DX2, leading to its subsequent degradation by the proteasome. mdpi.comnih.govbiorxiv.org In lung cancer cells with high expression of AIMP2-DX2, this compound potently suppresses cell growth. mdpi.com This targeted degradation of a pro-tumorigenic protein represents a specific and potent mechanism of this compound's antitumor activity, suggesting its potential as a therapeutic agent for lung cancers characterized by high levels of AIMP2-DX2. mdpi.comnih.gov
Suppression of NRF2 through DHFR Inhibition and One-Carbon Metabolism
This compound has been identified as an indirect inhibitor of the Nuclear factor erythroid 2-related factor 2 (NRF2) signaling pathway. nih.gov Its mechanism of action is not through direct binding to NRF2 or its negative regulator, Kelch-like ECH-associated protein 1 (KEAP1), but rather through the inhibition of dihydrofolate reductase (DHFR). nih.govmdpi.com DHFR is a crucial enzyme in one-carbon metabolism, responsible for regenerating tetrahydrofolate from dihydrofolate. cellsignal.com Tetrahydrofolate is essential for the synthesis of purines and pyrimidines, which are the building blocks of DNA and RNA. mdpi.comcellsignal.com
By inhibiting human DHFR (hDHFR), this compound disrupts the one-carbon transfer reactions necessary for nucleotide synthesis. nih.govmdpi.com This disruption of one-carbon metabolism is the key to its NRF2-suppressive activity. nih.gov Studies have shown that the inhibition of DHFR is a prerequisite for the suppression of NRF2 by this compound. nih.gov Metabolomic analyses have revealed strong similarities between the effects of this compound, its potent analogue WCDD115, and the classical DHFR inhibitor methotrexate. nih.gov Mechanistically, this compound has been shown to reduce the half-life of the NRF2 protein by promoting its ubiquitination and subsequent degradation, a process that occurs independently of KEAP1. researchgate.net This highlights the critical role of one-carbon metabolism in maintaining the stability and function of the NRF2 signaling pathway. nih.gov The suppression of NRF2 and its antioxidant functions represents a therapeutic strategy for cancers where NRF2 is overactive. nih.govmdpi.com
Table 1: Comparative Inhibitory Activity of this compound and Analogue WCDD115
| Compound | NRF2 Inhibition (IC₅₀) | hDHFR Inhibition (IC₅₀) | Potency vs. This compound (NRF2) | Potency vs. This compound (hDHFR) |
|---|---|---|---|---|
| This compound (PYR) | 1.2 µM | 4.49 µM | - | - |
| WCDD115 | 57 nM | 144 nM | 22-fold greater | 31-fold greater |
Data sourced from Chembo et al., 2025. nih.gov
Mitochondrial Dysfunction and Cytochrome c Release
This compound can induce apoptosis in certain cells through a mechanism involving direct action on mitochondria. researchgate.netmedicinacomplementar.com.br A key event in this process is the disruption of the mitochondrial membrane potential (MMP). researchgate.netmedicinacomplementar.com.br Studies using the A549 non-small cell lung cancer cell line demonstrated that this compound treatment leads to a reduction in MMP. researchgate.net This depolarization of the mitochondrial membrane is a critical step that precedes the release of pro-apoptotic factors from the mitochondrial intermembrane space into the cytosol. researchgate.netembopress.org
One of the most significant of these factors is cytochrome c. embopress.org Following the loss of MMP, this compound enhances the release of cytochrome c into the cytosol. researchgate.netresearchgate.net Once in the cytosol, cytochrome c participates in the formation of the apoptosome, a protein complex that activates downstream caspases and executes the apoptotic program. embopress.org The release of cytochrome c is therefore a hallmark of mitochondrially-driven apoptosis and is a direct consequence of this compound-induced mitochondrial dysfunction. researchgate.netresearchgate.net This pathway highlights a mechanism of action for this compound that is central to programmed cell death. researchgate.net
Table 2: Effects of this compound on Mitochondrial Apoptotic Events in A549 Cells
| Parameter | Observation | Implication |
|---|---|---|
| Mitochondrial Membrane Potential (MMP) | Reduced after 48 hours of treatment. researchgate.net | Indicates mitochondrial depolarization and dysfunction. |
| Cytochrome c | Enhanced release into the cytosol. researchgate.net | Triggers downstream apoptotic signaling cascade. |
Data sourced from Lin et al., 2018. researchgate.net
Immunomodulatory Activities, including Apoptosis in Peripheral Blood Lymphocytes
Beyond its antiparasitic functions, this compound possesses immunomodulatory properties, notably the ability to induce apoptosis in peripheral blood lymphocytes (PBLs). nih.govresearchgate.net This effect has been observed in both resting and activated T lymphocytes. researchgate.netresearchgate.net The induction of apoptosis in these immune cells is a key aspect of its immunomodulating activity. medicinacomplementar.com.br
The mechanism underlying this apoptosis involves the intrinsic pathway, characterized by direct effects on mitochondria, rather than relying on death receptors like CD95/Fas. medicinacomplementar.com.brresearchgate.net this compound treatment leads to the activation of upstream caspases, specifically caspase-8 and caspase-10, which subsequently activate effector caspases like caspase-3. researchgate.net Furthermore, this compound has been shown to down-regulate the expression of Bcl-2, an anti-apoptotic protein that normally functions to protect mitochondrial integrity. medicinacomplementar.com.brresearchgate.netresearchgate.net The reduction in Bcl-2 levels facilitates mitochondrial membrane depolarization and the release of cytochrome c, thereby promoting the apoptotic cascade. researchgate.net Studies have shown that in activated PBLs, the apoptotic effect of this compound is dose-dependent. researchgate.net In the context of malarial infections, this compound treatment has been found to increase apoptosis in B lymphocytes and modulate the expression of cytokines, such as upregulating interferon-gamma (IFN-γ) and transforming growth factor-beta (TGF-β). nih.gov
Table 3: Summary of this compound's Apoptotic Effects on Peripheral Blood Lymphocytes
| Molecular Target/Pathway | Effect of this compound | Reference |
|---|---|---|
| Caspase-8 and Caspase-10 | Activation. | researchgate.net |
| Bcl-2 Protein | Down-regulation of expression. | researchgate.netresearchgate.net |
| CD95/Fas Receptor | Apoptosis induction is independent of this pathway. | researchgate.net |
| B Lymphocytes | Increased apoptosis during P. yoelii infection. | nih.gov |
| Cytokine Expression (in vivo) | Upregulation of IFN-γ and TGF-β mRNA. | nih.gov |
Antiprotozoal Research and Resistance Mechanisms
Research on Plasmodium Species (Malaria)
Pyrimethamine's primary application in antiprotozoal research has been in the context of malaria. It targets a fundamental metabolic pathway in the parasite, which has also made it a focal point for understanding how parasites develop resistance to chemotherapy.
This compound functions as a potent antifolate agent, specifically targeting the asexual erythrocytic stages of the Plasmodium parasite, which are responsible for clinical malaria. nih.govasm.org It is a slow-acting blood schizonticidal drug. nih.gov Its mechanism of action is based on the differential requirement for nucleic acid precursors between the parasite and its human host. pediatriconcall.comdrugbank.com
The compound inhibits the enzyme dihydrofolate reductase (DHFR), a critical component of the folate biosynthesis pathway. patsnap.comfda.govdovepress.com DHFR is responsible for the conversion of dihydrofolate to tetrahydrofolate, a co-factor essential for the synthesis of purines and pyrimidines, which are the building blocks of DNA and RNA. drugbank.compatsnap.com By competitively binding to the active site of the parasite's DHFR with high affinity, this compound blocks this conversion, leading to a depletion of essential folate derivatives. patsnap.com This disruption of nucleic acid and protein synthesis ultimately leads to the failure of nuclear division during schizont formation and results in parasite death. drugbank.com this compound exhibits high selectivity for the plasmodial DHFR over the human equivalent, which is a key factor in its therapeutic use. pediatriconcall.compatsnap.com While it is highly effective against the asexual blood stages, it does not destroy gametocytes but can arrest their development (sporogony) in the mosquito vector. pediatriconcall.com
The widespread use of this compound has unfortunately led to the emergence and spread of resistant Plasmodium falciparum strains, particularly in regions like Southeast Asia, South America, and parts of Africa. fda.gov This resistance is primarily linked to specific genetic alterations in the parasite.
Resistance to this compound in P. falciparum is predominantly caused by the accumulation of point mutations in the gene encoding dihydrofolate reductase (Pfdhfr). plos.orgparahostdis.org These mutations alter the enzyme's structure, reducing the binding affinity of this compound to its target.
Molecular Basis of this compound Resistance in Plasmodium falciparum
Point Mutations in P. falciparum Dihydrofolate Reductase (Pfdhfr) Gene
Specific Amino Acid Substitutions and Their Impact on Binding Affinity
Several key amino acid substitutions in the Pfdhfr gene have been identified as direct contributors to this compound resistance. The foundational mutation is typically a change from serine to asparagine at codon 108 (S108N). asm.orgird.frird.frtandfonline.com This single mutation is strongly associated with in vitro resistance to this compound. ird.frnih.gov The S108N mutation is thought to cause steric hindrance with the chlorine atom of this compound's aryl group, thereby reducing the drug's binding affinity. researchgate.net
Additional mutations can build upon the S108N background to confer higher levels of resistance. These include:
N51I (Asparagine to Isoleucine at codon 51) : This mutation, in conjunction with S108N, further increases resistance. asm.orgasm.org
C59R (Cysteine to Arginine at codon 59) : Similar to N51I, the C59R mutation adds to the level of resistance when present with other mutations. asm.orgasm.org
I164L (Isoleucine to Leucine at codon 164) : The I164L mutation, when combined with others, leads to a very high level of this compound resistance and can significantly compromise the clinical efficacy of drugs containing it. ird.frasm.org
The binding affinity of this compound is progressively reduced as these mutations accumulate. pnas.org For instance, parasites with a single S108N mutation show a significant increase in resistance, and this is further amplified by the addition of N51I or C59R mutations. asm.org
Table 1: Key Amino Acid Substitutions in Pfdhfr and their Impact on this compound Resistance
| Mutation | Amino Acid Change | Impact on Resistance Level |
| S108N/T | Serine to Asparagine/Threonine | Foundational mutation for this compound resistance. asm.orgspringermedizin.de |
| N51I | Asparagine to Isoleucine | Confers a higher level of resistance in combination with S108N. asm.org |
| C59R | Cysteine to Arginine | Further increases resistance when present with other mutations. asm.org |
| I164L | Isoleucine to Leucine | Associated with very high levels of resistance, especially in quadruple mutants. ird.frasm.org |
Accumulation of Multiple Mutations and Exacerbated Resistance
The development of high-level this compound resistance is a stepwise process involving the accumulation of multiple mutations in the Pfdhfr gene. springermedizin.de While the S108N mutation alone confers a moderate level of resistance, the presence of double, triple, or quadruple mutations leads to a significant increase in the parasite's ability to withstand the drug. asm.orgird.fr
The "triple mutant" (N51I, C59R, and S108N) is strongly associated with a high degree of this compound resistance and is a reliable predictor of treatment failure in vivo. asm.orgmdpi.com The addition of the I164L mutation to this combination, creating a "quadruple mutant," results in even higher levels of resistance, often rendering this compound-based therapies ineffective. ird.frasm.org Studies have shown a direct correlation between the number of Pfdhfr point mutations and the level of both in vitro and in vivo resistance to this compound. asm.org
This compound is frequently used in combination with sulfadoxine, a drug that inhibits a different enzyme in the same folate pathway, dihydropteroate synthase (DHPS). fda.govnih.gov This combination is designed to be synergistic, meaning its combined effect is greater than the sum of its individual effects. oup.comsci-hub.se However, resistance can also develop to this combination therapy.
The synergistic action of the two drugs means that resistance often requires mutations in both target genes: Pfdhfr for this compound and Pfdhps (dihydropteroate synthase gene) for sulfadoxine. oup.com Point mutations in the Pfdhps gene, such as A437G and K540E, are associated with sulfadoxine resistance. asm.orgspringermedizin.de
The presence of mutations in both genes leads to synergistic resistance. For example, a parasite with triple mutations in Pfdhfr (conferring high-level this compound resistance) combined with double mutations in Pfdhps (the "quintuple mutant") is strongly associated with treatment failure of the sulfadoxine-pyrimethamine combination. plos.orgspringermedizin.de The accumulation of mutations appears to follow an ordered pathway, often with initial mutations occurring in Pfdhfr before accumulating in Pfdhps. oup.com This combined resistance undermines the efficacy of the synergistic drug combination. asm.orgasm.org
Molecular Epidemiology of this compound Resistance
The emergence and spread of this compound resistance in Plasmodium falciparum, the parasite responsible for the most severe form of malaria, is a significant public health concern. This resistance is primarily associated with specific point mutations in the parasite's dihydrofolate reductase (dhfr) gene, the target of this compound.
This compound resistance has been observed to originate in specific focal points and subsequently spread globally. oup.com Highly resistant parasite strains, characterized by three or more mutations in the dhfr gene, are thought to have emerged from a limited number of geographical locations, possibly one in Asia and two in South America, before disseminating worldwide. oup.com The Thailand-Cambodia border is considered a primary epicenter for the origin of this compound resistance. nih.gov From there, resistant alleles have migrated to other parts of Southeast Asia and Africa, following a pattern similar to that of chloroquine resistance. oup.comnih.gov
The prevalence of specific this compound resistance-conferring mutations varies geographically. In Central Africa, a high prevalence of Pfdhfr single-nucleotide polymorphisms (SNPs) has been observed, with the triple mutation haplotype (I51R59N108) being particularly frequent in countries like Cameroon, Equatorial Guinea, and Gabon. malariaworld.org Studies in southern China have revealed distinct distribution patterns of pfdhfr alleles between the Yunnan-Burma border and Hainan Island, reflecting their different histories of drug administration. nih.gov
In eastern Africa, there has been a significant increase in mid-level resistance to sulfadoxine-pyrimethamine (SP), a combination therapy, in several countries, including Tanzania, Sudan, Mozambique, and Kenya. bmj.com The prevalence of the pfdhps 540E mutation, a marker for SP resistance, has expanded from limited foci in East Africa to exceed 50% in most of East and South East Africa. plos.org While western Africa has generally maintained low levels of resistance, some countries like Mali have seen an increase. bmj.com
The global migration of these resistant parasites has been a decisive factor in the establishment of drug-resistant P. falciparum populations. oup.com
Longitudinal studies are crucial for monitoring the evolution of this compound resistance over time. In Yaoundé, Cameroon, surveillance from 1994 to 2001 showed a significant and progressive increase in the proportion of this compound-resistant P. falciparum isolates. ird.fr The prevalence of isolates with mutant dhfr alleles rose from 42–45% in 1994–1995 to 88–92% in 2000–2001, with a predominance of triple dhfr mutations in the later years. ird.fr
In Maputo Province, Mozambique, annual surveillance between 2004 and 2008, following the implementation of artesunate plus SP, revealed that the dhfr triple mutation remained close to fixation. lshtm.ac.uk Furthermore, the "quintuple" mutation (dhfr triple mutant combined with dhps double mutant) increased dramatically from 11.0% in 2004 to 75.0% by 2008. lshtm.ac.uk
Conversely, in regions where SP drug pressure has been removed, a decline in resistance markers has been observed. In northwestern Ethiopia, after the withdrawal of SP, there was a significant decrease in the frequency of the triple Pfdhfr mutation from 50.8% to 15.9%. plos.org This suggests that in the absence of drug pressure, resistant strains may be less fit and are gradually replaced by sensitive ones. plos.org However, other long-term studies, such as one in Tanganyika (now part of Tanzania), showed that this compound-resistant parasites could still be detected 20 years after the initial observations of their decline in the absence of drug pressure. who.int
A study in Kilifi, Kenya, revealed that mutations in dhfr associated with this compound resistance were present at high frequencies even before the widespread use of the drug and remained high after its replacement with another first-line treatment. wellcomeopenresearch.org This highlights the complexity of resistance dynamics and the persistence of resistance mutations in a population.
A strong correlation exists between the presence of specific mutations in the dhfr gene and the in vitro sensitivity of P. falciparum to this compound. The 50% inhibitory concentration (IC50), a measure of drug effectiveness in vitro, increases with the number of dhfr mutations.
Studies have consistently shown that isolates with wild-type dhfr alleles are sensitive to this compound, exhibiting low IC50 values. ird.frasm.org In contrast, isolates with single, double, or triple mutations in the dhfr gene show progressively higher IC50 values, indicating increased resistance. ird.frasm.org
For instance, Kenyan field isolates could be categorized into three distinct groups based on their this compound IC50s and dhfr genotypes. asm.org Wild-type isolates had the lowest IC50s, while those with mutations at codon 108 (with or without mutations at codons 51 or 59) showed a significant increase in resistance. asm.org The highest level of resistance was observed in parasites with the triple mutation (Ile-51, Arg-59, and Asn-108). asm.org
Similarly, in Cameroon, a clear distinction was observed between isolates with pure wild-type dhfr alleles (IC50 < 100 nM) and those with pure mutant alleles (IC50 ≥ 100 nM). ird.fr This high correlation between in vitro results and molecular markers validates the use of molecular surveillance to predict this compound resistance levels in a population. ird.fr
The presence of the dhfr triple mutant has been suggested as a useful predictor of treatment failure in some studies. ajtmh.org However, the predictive value can vary regionally. researchgate.net In some regions, the combination of the dhfr triple mutant and specific dhps mutations (such as the Gly-437 + Glu-540 double mutant) is a better predictor of SP treatment failure. ajtmh.org
Table 1: Correlation of P. falciparum dhfr Genotype with In Vitro this compound Resistance
| dhfr Genotype | Number of Mutations | This compound IC50 Range/Mean (nM) | Resistance Level | Reference |
| Wild-type | 0 | Mean: 3.71 | Sensitive | asm.org |
| Asn-108 (single) or with Ile-51 or Arg-59 (double) | 1-2 | - | Intermediate Resistance | asm.org |
| Ile-51 + Arg-59 + Asn-108 (triple) | 3 | Mean: 815.25 | High Resistance | asm.org |
| Wild-type | 0 | < 100 | Sensitive | ird.fr |
| Single, double, or triple mutations | 1-3 | ≥ 100 | Resistant | ird.fr |
Longitudinal Surveillance of Resistance Trends
Strategies to Circumvent this compound Resistance
The widespread resistance to this compound has necessitated the development of strategies to overcome this challenge. A primary approach is the use of combination therapies. patsnap.comtandfonline.com Combining this compound with other antimalarial drugs that have different mechanisms of action can enhance efficacy and delay the development of resistance. patsnap.com The most well-known combination is sulfadoxine-pyrimethamine (SP), where sulfadoxine inhibits another enzyme in the folate pathway, dihydropteroate synthase (DHPS). patsnap.com
Another strategy is the development of novel antifolates that are effective against this compound-resistant strains. mdpi.comoup.com P218 is a new dihydrofolate reductase (DHFR) inhibitor that has shown high selectivity and activity against this compound-resistant P. falciparum. mdpi.combiorxiv.org It has demonstrated potent inhibition of both the asexual and sexual stages of this compound-resistant parasites. biorxiv.orgresearchgate.net
The combination of a short-acting antifolate, such as chlorproguanil, with dapsone has also been explored as an alternative to SP. oup.com This combination is thought to select less efficiently for resistance due to the shorter half-life of the drugs. oup.com
Furthermore, the use of triple combination therapies is being investigated. tandfonline.com However, the effectiveness of this approach may be limited if significant resistance to the component drugs already exists. tandfonline.com
In some regions where this compound resistance is high, the drug has been replaced by artemisinin-based combination therapies (ACTs) as the first-line treatment for uncomplicated malaria.
Research on Toxoplasma gondii (Toxoplasmosis)
Toxoplasma gondii is an obligate intracellular protozoan parasite that causes toxoplasmosis. medscape.com this compound is a key component in the treatment of this infection. medscape.comcdc.gov
This compound's Role in Inhibition of T. gondii Growth
This compound effectively inhibits the growth of Toxoplasma gondii by targeting a crucial metabolic pathway. patsnap.comnih.gov The parasite, like Plasmodium, relies on the synthesis of folic acid for its survival and replication. patsnap.comnih.gov
The mechanism of action of this compound against T. gondii is the inhibition of the enzyme dihydrofolate reductase (DHFR). patsnap.comnih.govoup.com DHFR is essential for the conversion of dihydrofolate to tetrahydrofolate, a vital cofactor in the synthesis of nucleic acids (DNA and RNA) and certain amino acids. patsnap.com By blocking DHFR, this compound disrupts these synthetic processes, leading to an inhibition of parasite growth and ultimately cell death. patsnap.comdrugbank.com
This compound exhibits a higher affinity for the Toxoplasma DHFR enzyme compared to the mammalian host's enzyme, which contributes to its therapeutic effect. oup.com However, this selectivity is not absolute. patsnap.com
In clinical practice, this compound is often used in combination with a sulfonamide, such as sulfadiazine. patsnap.com This combination provides a synergistic effect because sulfadiazine inhibits dihydropteroate synthase (DHPS), another enzyme in the folate biosynthesis pathway. nih.govasm.org The simultaneous blockage of two steps in this pathway is highly effective at inhibiting the proliferation of T. gondii. nih.gov Studies have shown that combining this compound with other drugs can significantly reduce the parasite load in experimental models. brieflands.com In vitro studies have demonstrated that the addition of even low concentrations of this compound can dramatically increase the inhibitory effect of sulfamethoxazole. oup.com
Mechanisms of Resistance in T. gondii to this compound
Resistance to this compound in the protozoan parasite Toxoplasma gondii is a significant concern in the management of toxoplasmosis. The primary mechanism of this resistance involves specific genetic mutations in the parasite's dihydrofolate reductase-thymidylate synthase (DHFR-TS) gene. nih.govfrontiersin.org this compound functions by inhibiting the DHFR enzyme, which is crucial for the synthesis of nucleic acids and some amino acids, thereby halting parasite replication. frontiersin.orgpatsnap.com
Research has identified that point mutations within the DHFR coding region can significantly reduce the binding affinity of this compound to the enzyme, allowing the parasite to continue its metabolic functions even in the presence of the drug. patsnap.com These mutations are analogous to those observed in the malaria parasite, Plasmodium falciparum, which also exhibits resistance to this compound. nih.govpnas.org
Several key amino acid substitutions in the T. gondii DHFR enzyme have been identified as conferring resistance. In vitro studies involving random mutagenesis have pinpointed mutations at various residues. For instance, substitutions such as Tryptophan to Arginine at position 25 (W25R), Leucine to Serine at position 98 (L98S), and Leucine to Hisitidine at position 134 (L134H) have been shown to produce drug resistance. nih.govasm.orgnih.govfrontiersin.org
Furthermore, site-directed mutagenesis experiments have revealed other critical mutations. A T83N mutation (Threonine to Asparagine at codon 83) has been found to likely confer resistance to this compound. frontiersin.org The level of resistance can be further amplified when multiple mutations are present. For example, the T83N mutation, when combined with S36R (Serine to Arginine at codon 36) and F245S (Phenylalanine to Serine at codon 245), results in increased resistance. frontiersin.org The accumulation of multiple mutations in the DHFR-TS gene is associated with high-level resistance. nih.gov
It is important to note that different strains of T. gondii may exhibit varying intrinsic sensitivities to this compound, even without mutations in the predicted DHFR-TS protein sequences. nih.govasm.org While the primary focus of resistance research has been on target site modifications, the adaptive potential of the T. gondii parasite suggests that other mechanisms may also contribute to drug resistance, although these are not as well understood. nih.gov
Pyrimethamine in Anticancer Research
Preclinical Efficacy Studies in Cancer Models
Preclinical studies using both cancer cell lines and animal models have been instrumental in elucidating the anticancer properties of pyrimethamine. These investigations have shown promising results in controlling cancer cell proliferation, triggering cell death, and hindering the advancement of tumors.
Effects on Cell Proliferation and Viability in Various Cancer Cell Lines
This compound has been shown to effectively reduce the viability of a wide range of cancer cell lines in a dose-dependent manner. nih.govaacrjournals.org For instance, in non-small cell lung cancer (NSCLC) A549 cells, this compound treatment led to a gradual decrease in cell viability with increasing concentrations and longer exposure times. iiarjournals.org This was accompanied by morphological changes indicative of cell distress, such as shrinkage and rounding. iiarjournals.org
Similarly, studies on colorectal cancer (CRC) cell lines, including HCT116, DLD1, RKO, SW480, and HT29, demonstrated that this compound significantly inhibited their growth. nih.gov The inhibitory effect of this compound has also been observed in ovarian cancer cell lines (SKOV3, A2780, OVCAR8, and ES2), where it suppressed cell viability with IC50 values ranging from 20 to 60 µM. techscience.com In prostate cancer, this compound has been found to inhibit the proliferation of DU145 and PC3 cells by inducing S-phase cell cycle arrest. nih.gov
The mechanism behind this anti-proliferative effect is often linked to the inhibition of dihydrofolate reductase (DHFR), an enzyme crucial for DNA synthesis. nih.govaacrjournals.org By blocking this enzyme, this compound disrupts the production of nucleic acids, thereby halting the proliferation of rapidly dividing cancer cells. iiarjournals.orgtechscience.com Some research suggests that the sensitivity of cancer cells to this compound may be correlated with the expression levels of DHFR. aacrjournals.org
| Cancer Type | Cell Lines | Observed Effects | Key Findings |
|---|---|---|---|
| Non-Small Cell Lung Cancer | A549, NCI-H460, NCI-H446 | Reduced cell viability, morphological changes. aacrjournals.orgiiarjournals.org | Inhibition of DHFR activity. aacrjournals.org |
| Colorectal Cancer | HCT116, DLD1, RKO, SW480, HT29 | Dose-dependent growth inhibition. nih.gov | Induction of S-phase arrest. nih.gov |
| Ovarian Cancer | SKOV3, A2780, OVCAR8, ES2 | Suppressed cell viability (IC50: 20-60 µM). techscience.com | Inhibition of cell proliferation. techscience.com |
| Prostate Cancer | DU145, PC3 | Inhibited proliferation, S-phase arrest. nih.gov | Suppression of DNA synthesis. |
| Breast Cancer | MCF-7 | Inhibitory activity observed. aacrjournals.org | Targeting of DHFR. aacrjournals.org |
Induction of Apoptosis in Cancer Cells
A significant aspect of this compound's anticancer activity is its ability to induce apoptosis, or programmed cell death, in various cancer cell types. researchgate.netiiarjournals.org This process is a key mechanism for eliminating malignant cells.
In non-small cell lung cancer (NSCLC) A549 cells, this compound treatment led to an increase in sub-G1 phase accumulation, a hallmark of apoptosis. iiarjournals.org This was accompanied by the activation of key apoptotic proteins, caspase-9 and caspase-3, and the cleavage of poly (ADP-ribose) polymerase (PARP). iiarjournals.org Furthermore, this compound induced mitochondrial dysfunction, as evidenced by a reduction in mitochondrial membrane potential and the release of cytochrome c. iiarjournals.org The study also noted a decrease in the anti-apoptotic proteins Bcl-2 and Bcl-xL, while the pro-apoptotic protein Bak was significantly increased. iiarjournals.org These findings suggest that this compound triggers the intrinsic, mitochondria-mediated apoptotic pathway in NSCLC cells. iiarjournals.org
Similarly, in castration-resistant prostate cancer (CRPC) cell lines, DU145 and PC3, this compound was shown to promote apoptosis. researchgate.net Flow cytometry analysis revealed a significant increase in apoptotic cells following treatment. researchgate.net This was further supported by a decrease in the expression of the anti-apoptotic protein Bcl-2 and an increase in the cleavage of caspase-3. researchgate.net The pro-apoptotic effects in prostate cancer cells are thought to be mediated through the inhibition of the p38/NF-κB signaling pathway. researchgate.net
Research on melanoma cells has also demonstrated that this compound can induce apoptosis through a mechanism involving both caspases and cathepsins. iiarjournals.orgoup.com In some cancer types, such as ovarian cancer, apoptosis was observed at later time points or higher concentrations, suggesting it may not be the primary mode of cell death in all cases. techscience.com
| Cancer Type | Cell Lines | Key Apoptotic Events | Signaling Pathways Implicated |
|---|---|---|---|
| Non-Small Cell Lung Cancer | A549 | Activation of caspase-9 and -3, PARP cleavage, mitochondrial dysfunction. iiarjournals.org | Intrinsic (mitochondrial-mediated) pathway. iiarjournals.org |
| Prostate Cancer | DU145, PC3 | Decreased Bcl-2, increased cleaved caspase-3. researchgate.net | Inhibition of p38/NF-κB pathway. researchgate.net |
| Melanoma | Not specified | Caspase and cathepsin activation. iiarjournals.orgoup.com | Not specified |
| Ovarian Cancer | A2780s, SKOV3, ES2, OVCAR8 | Delayed caspase-3 cleavage. techscience.com | Apoptosis is not the primary cell death mechanism. techscience.com |
Suppression of Tumor Growth in Xenograft Models
The antitumor effects of this compound observed in cell cultures have been successfully translated to in vivo animal models. Xenograft studies, where human cancer cells are implanted into immunodeficient mice, have demonstrated this compound's ability to significantly suppress tumor growth.
In a xenograft model using non-small cell lung cancer A549 cells, mice treated with this compound showed a notable reduction in tumor growth compared to the control group. iiarjournals.org After 35 days, the tumors in the this compound-treated mice were significantly smaller and weighed less. iiarjournals.org Similarly, in a Lewis lung cancer (LLC) xenograft model, this compound treatment suppressed tumor growth. aacrjournals.org
Research on prostate cancer has also yielded positive results. In a xenograft model using PC3 prostate cancer cells, oral administration of this compound led to a substantial reduction in tumorigenesis. researchgate.net This was accompanied by a decrease in the expression of Ki-67, a marker of cell proliferation, and phosphorylated p38 in the tumor tissues. researchgate.net Another study using H460 lung cancer cells in a mouse xenograft model showed that this compound significantly reduced both the size and weight of tumors, with an efficacy comparable to the chemotherapy drug taxol. mdpi.com
Furthermore, in a melanoma xenograft model, this compound treatment initiated after the onset of metastatic tumors resulted in a significant reduction in tumor weight over time. researchgate.net These findings from various xenograft models provide strong evidence for the in vivo antitumor efficacy of this compound. researchgate.netiiarjournals.orgnih.gov
| Cancer Type | Cell Line Used | Animal Model | Key Findings |
|---|---|---|---|
| Non-Small Cell Lung Cancer | A549 | SCID mice | Reduced tumor growth, smaller and lighter tumors. iiarjournals.org |
| Lung Cancer | Lewis Lung Cancer (LLC) | C57BL/6J mice | Suppressed tumor growth. aacrjournals.org |
| Prostate Cancer | PC3 | Nude mice | Substantial reduction in tumorigenesis, decreased Ki-67 and phosphorylated p38 expression. researchgate.net |
| Lung Cancer | H460 | BALB/cSLC-nu/nu mice | Significantly reduced tumor size and weight, comparable to taxol. mdpi.com |
| Melanoma | 501 | SCID mice | Significant reduction in tumor weight. researchgate.net |
Inhibition of Metastasis and Invasion (e.g., Epithelial-Mesenchymal Transition)
Beyond its effects on cell growth and survival, this compound has been shown to inhibit the processes of metastasis and invasion, which are critical for cancer progression and spread. A key mechanism involved in this is the inhibition of the epithelial-mesenchymal transition (EMT). aacrjournals.orgnih.gov EMT is a process where epithelial cells lose their characteristics and gain mesenchymal features, enabling them to become more motile and invasive. mdpi.com
In lung cancer cells, while both this compound and the standard chemotherapy drug methotrexate can inhibit cell proliferation, only this compound was found to inhibit EMT, migration, and invasion. aacrjournals.orgnih.gov This suggests that this compound has an additional target beyond DHFR that is involved in these processes. aacrjournals.orgnih.gov Research has identified thymidine phosphorylase (TP) as another target of this compound. aacrjournals.orgnih.gov TP is an enzyme associated with the EMT of cancer cells. aacrjournals.org By targeting both DHFR and TP, this compound plays a dual role in inhibiting both tumor proliferation and metastasis. aacrjournals.orgnih.gov
Studies on lung cancer cell lines NCI-H460 and A549 demonstrated that this compound inhibited cell migration and invasion. aacrjournals.org Western blot analysis revealed that this compound treatment led to changes in the expression of EMT markers, such as an increase in E-cadherin (an epithelial marker) and a decrease in vimentin (a mesenchymal marker). aacrjournals.org In vivo studies using a Lewis lung cancer xenograft model further confirmed that this compound treatment drastically decreased the number of metastatic tumor nodes on the lung surfaces. aacrjournals.org
Specific Cancer Types Under Investigation
The anticancer potential of this compound is being explored in a variety of malignancies. One area of significant focus has been prostate cancer.
Prostate Cancer
This compound has demonstrated significant antitumor effects in preclinical models of prostate cancer, particularly in castration-resistant prostate cancer (CRPC), a more advanced and difficult-to-treat form of the disease. researchgate.netnih.gov
Studies on the metastatic prostate cancer cell lines DU145 and PC3 have shown that this compound inhibits cell proliferation, induces cell cycle arrest in the S phase, and promotes apoptosis. nih.gov The inhibition of cell proliferation is linked to the suppression of DNA synthesis. The pro-apoptotic effect is evidenced by a decrease in the anti-apoptotic protein Bcl-2 and an increase in cleaved caspase-3. researchgate.net
The mechanism of action in prostate cancer has been linked to the inhibition of the p38-NF-κB signaling pathway. researchgate.netnih.gov In vivo studies using a xenograft model with PC3 cells showed that oral administration of this compound suppressed tumor growth. nih.gov This was associated with reduced expression of the proliferation marker Ki-67 and phosphorylated p38 in the tumors. researchgate.net
Furthermore, this compound has been shown to reduce the activity of telomerase, an enzyme important for cancer cell immortality, in PC3 cells. researchgate.net These findings highlight the multifaceted ways in which this compound exerts its antitumor effects on prostate cancer, suggesting its potential as a novel therapeutic strategy for CRPC. nih.gov
Lung Cancer (e.g., Non-Small Cell Lung Carcinoma)
This compound has demonstrated notable antitumor effects in non-small cell lung cancer (NSCLC). iiarjournals.orgiiarjournals.org Its primary mechanism involves the inhibition of dihydrofolate reductase (DHFR), an enzyme essential for DNA synthesis. aacrjournals.orgdrugbank.comwikipedia.org By blocking DHFR, this compound disrupts the folate metabolism, leading to cell cycle arrest and apoptosis (programmed cell death). wikipedia.orgnih.govnih.gov
In vitro studies on the A549 human NSCLC cell line have shown that this compound reduces cell viability and induces G0/G1 phase arrest in the cell cycle. iiarjournals.orgnih.gov This is associated with the downregulation of key cell cycle proteins such as cyclins D1 and E, as well as cyclin-dependent kinases (CDK) 4 and 2, and the upregulation of the cell cycle inhibitor p21. iiarjournals.orgnih.gov Furthermore, this compound treatment leads to an accumulation of cells in the sub-G1 phase, a hallmark of apoptosis, and triggers mitochondrial dysfunction. iiarjournals.orgiiarjournals.org This is evidenced by the activation of caspases-9 and -3 and the cleavage of poly (ADP-ribose) polymerase (PARP). iiarjournals.orgnih.gov
Another significant finding is this compound's ability to inhibit the AIMP2-DX2 protein, a splice variant of AIMP2 that is highly expressed in lung cancer and its levels increase with tumor progression. mdpi.com this compound induces the ubiquitin-mediated degradation of AIMP2-DX2, thereby suppressing tumor growth. mdpi.com In a mouse xenograft model using H460 lung cancer cells, which have high levels of AIMP2-DX2, this compound administration significantly reduced tumor size and weight, comparable to the effects of the chemotherapy drug taxol. mdpi.com
Moreover, research has identified thymidine phosphorylase (TP) as another target of this compound in lung cancer cells. aacrjournals.orgnih.gov While both this compound and the standard chemotherapy drug methotrexate inhibit DHFR, only this compound was found to also inhibit the epithelial-mesenchymal transition (EMT), metastasis, and invasion of lung cancer cells, suggesting a dual targeting mechanism involving both DHFR and TP. aacrjournals.orgnih.gov In vivo studies using a Lewis lung cancer xenograft model in mice confirmed that this compound suppressed tumor growth and drastically decreased the number of metastatic tumor nodes on the lung surfaces. aacrjournals.org
Table 1: Research Findings of this compound in Lung Cancer
| Model System | Key Findings | Mechanism of Action | Reference |
|---|---|---|---|
| A549 NSCLC cell line | Reduced cell viability, induced G0/G1 arrest, induced apoptosis. | Down-regulation of cyclin D1, cyclin E, CDK4, CDK2; up-regulation of p21; activation of caspase-9, caspase-3. | iiarjournals.orgnih.gov |
| H460 lung cancer cells | Suppressed cell growth, reduced tumor size and weight in vivo. | Induced ubiquitin-mediated degradation of AIMP2-DX2. | mdpi.com |
| NCI-H460 & A549 cells | Inhibited cell proliferation, migration, and invasion. | Dual inhibition of dihydrofolate reductase (DHFR) and thymidine phosphorylase (TP). | aacrjournals.orgnih.gov |
| A549 xenograft model | Effectively inhibited NSCLC tumor growth. | Induced G1 cell-cycle arrest and intrinsic apoptotic pathway. | iiarjournals.orgresearchgate.net |
Melanoma
In the context of melanoma, this compound has been shown to induce apoptosis in cancer cells through a dual mechanism involving both caspases and cathepsins. researchgate.net Studies have demonstrated that this compound treatment leads to a significant increase in the activities of caspases-3/7, -8, and -9 in melanoma cell lines. researchgate.net
Furthermore, this compound has been observed to work synergistically with the chemotherapy drug temozolomide, enhancing its cytotoxic effects on melanoma cells. nih.govnih.gov This suggests a potential for combination therapies to improve treatment outcomes for melanoma patients. Research on derivatives of this compound, such as methylbenzoprim, has also shown marked tumor regression in mouse models of melanoma. researchgate.net
Table 2: Research Findings of this compound in Melanoma
| Model System | Key Findings | Mechanism of Action | Reference |
|---|---|---|---|
| Melanoma cell lines | Induced apoptosis. | Activation of caspases and cathepsins. | researchgate.net |
| Melanoma cell lines | Enhanced temozolomide-induced cytotoxicity. | Synergistic effect with temozolomide. | nih.govnih.gov |
| SCID mouse model | Marked tumor regression. | Antitumor efficacy of a this compound derivative. | researchgate.net |
Breast Cancer
This compound has shown promise in preclinical models of breast cancer by inhibiting the STAT3 signaling pathway. nih.govoup.com STAT3 is a transcription factor that, when activated, promotes tumor cell proliferation, invasion, and migration. nih.govoup.com
In vitro studies using metastatic breast cancer cell lines demonstrated that this compound inhibited STAT3 activity, leading to reduced tumor cell proliferation and invasion. nih.gov In tumor-bearing mice, treatment with this compound resulted in reduced STAT3 activation within the tumor cells, attenuated tumor growth, and decreased tumor-associated inflammation. nih.gov An interesting finding was the enhanced cytotoxic activity of tumor-infiltrating CD8+ T cells, suggesting that this compound may also have an immune-stimulatory effect. nih.gov The potential therapeutic relevance of this compound may extend to triple-negative breast cancer, a subtype where STAT3 activation is known to support cancer stem cells. nih.gov
Table 3: Research Findings of this compound in Breast Cancer
| Model System | Key Findings | Mechanism of Action | Reference |
|---|---|---|---|
| TUBO and TM40D-MB metastatic breast cancer cells (in vitro) | Inhibited STAT3 activity, tumor cell proliferation, and invasion. | Inhibition of STAT3 signaling pathway. | nih.gov |
| Tumor-transplanted mice (in vivo) | Reduced STAT3 activation, attenuated tumor growth, reduced tumor-associated inflammation, enhanced CD8+ T cell cytotoxicity. | Direct and indirect tumor inhibitory effects through STAT3 inhibition. | nih.gov |
Acute Myeloid Leukemia
In the context of acute myeloid leukemia (AML), this compound has been identified as a potent inducer of apoptosis and differentiation in leukemia cells. nih.gov High-throughput drug screening identified this compound as an effective agent against AML cell lines, with its antileukemic effects partially attributed to its function as a dihydrofolate reductase (DHFR) antagonist. nih.gov
Oral administration of this compound was effective in two different xenograft mouse models of AML, where it specifically targeted leukemic cells while leaving healthy CD34+ cells unaffected. nih.govresearchgate.net The cytotoxic effects of this compound on AML cells could be partially reversed by the addition of excess folic acid, highlighting the importance of folate metabolism as a therapeutic target in this disease. nih.gov Further studies have shown that STAT3 is upregulated in the hematopoietic stem cells of MDS and AML patients, and its increased expression is associated with a poorer prognosis. ashpublications.org this compound, acting as a STAT3 inhibitor, has been shown to inhibit the proliferation of leukemic cell lines by inducing apoptosis. ashpublications.org
Table 4: Research Findings of this compound in Acute Myeloid Leukemia
| Model System | Key Findings | Mechanism of Action | Reference |
|---|---|---|---|
| Murine and human AML cell lines | Potent inducer of apoptosis and differentiation. | DHFR antagonist, targeting of folate metabolism. | nih.gov |
| Xenograft mouse models (HL60 and THP1 cells) | Significantly reduced tumor volumes. | Specific targeting of leukemic cells. | researchgate.netashpublications.org |
| Primary human AML cells | Significantly reduced colony numbers. | Specific targeting of leukemic cells. | researchgate.netashpublications.org |
| Leukemic cell lines (KG-1, KT-1, CMK) | Inhibited cellular proliferation by inducing apoptosis. | Inhibition of STAT3 activation. | ashpublications.org |
Pituitary Adenomas
Research has indicated that this compound can sensitize pituitary adenoma cells to the chemotherapeutic agent temozolomide (TMZ). nih.gov Invasive pituitary adenomas are often resistant to conventional therapies, making the synergistic effect of this compound and TMZ a promising therapeutic strategy. nih.gov
In vitro studies on various rat and mouse pituitary adenoma cell lines showed that the combination of this compound and TMZ synergistically inhibited cell proliferation and invasion, and induced apoptosis. nih.gov This combination treatment also led to cell cycle arrest and increased DNA damage. nih.gov The mechanism appears to involve the upregulation of cathepsin B and BAX, an increase in the activity of caspases-3/7, -8, and -9, and the release of cytochrome c from mitochondria. nih.gov In a xenograft tumor model using GH3 pituitary adenoma cells, the combination therapy produced synergistic antitumor activity and improved the survival rate of the animals without increasing systemic side effects. nih.gov
Table 5: Research Findings of this compound in Pituitary Adenomas
| Model System | Key Findings | Mechanism of Action | Reference |
|---|---|---|---|
| Rat/mouse PA cell lines (αT3-1, GH3, MMQ, ATt-20) | Synergistically inhibited proliferation, invasion, and induced apoptosis with TMZ. | Triggering of cathepsin B-dependent and caspase-dependent apoptotic pathways. | nih.gov |
| GH3 xenograft tumor model | Synergistic antitumor activity and enhanced survival rate with TMZ. | Increased DNA damage, upregulation of apoptotic proteins. | nih.gov |
Ovarian Cancer
This compound has been investigated for its potential therapeutic effects in ovarian cancer. mdpi.com It has been shown to inhibit the proliferation of ovarian cancer cells and reduce angiogenesis in mouse models. techscience.com The primary mechanism of action in ovarian cancer appears to be the induction of lethal mitophagy, a process of selective degradation of mitochondria by autophagy. techscience.com
In vitro studies have shown that this compound suppresses the proliferation of ovarian cancer cells. techscience.com While it does induce apoptosis, as indicated by the cleavage of caspase-3, this appears to be a secondary effect that occurs after prolonged exposure. techscience.com The more immediate and primary mechanism of cell death is lethal mitophagy, which is triggered by the activation of the p38/JNK/ERK signaling pathway. techscience.com These findings suggest that inducing mitochondrial autophagy could be an effective strategy for treating ovarian cancer. techscience.com
Table 6: Research Findings of this compound in Ovarian Cancer
| Model System | Key Findings | Mechanism of Action | Reference |
|---|---|---|---|
| Ovarian cancer cells (in vitro) | Suppressed cell proliferation. | Induction of lethal mitophagy via activation of the p38/JNK/ERK pathway. | techscience.com |
| Mouse models of ovarian cancer | Reduced angiogenesis, inhibited tumor cell proliferation. | Induction of lethal mitophagy. | techscience.com |
Triple-Negative Breast Cancer
The anticancer potential of this compound extends to triple-negative breast cancer (TNBC), a particularly aggressive subtype with limited treatment options. mdpi.com The inhibition of STAT3, a key mechanism of this compound, is highly relevant for TNBC as STAT3 activation is known to support the survival and proliferation of cancer stem cells in this subtype. nih.gov While direct studies on this compound in TNBC models are emerging, the established role of STAT3 in TNBC pathogenesis suggests that this compound could be a valuable therapeutic agent. mdpi.comnih.gov
Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia (CLL) is a type of cancer characterized by the excessive production of lymphocytes in the bone marrow. A key molecular feature of CLL is the constitutive activation of the STAT3 (Signal Transducer and Activator of Transcription 3) protein, which plays a crucial role in tumor cell growth. clinicaltrials.govashpublications.org Laboratory studies have demonstrated that this compound can induce the death of CLL cells. clinicaltrials.gov This has led to clinical trials aimed at evaluating its therapeutic potential in patients with this disease. clinicaltrials.govdana-farber.orgcancer.govclinicaltrials.gov
A phase I clinical trial was conducted to determine the safety and optimal dosage of this compound in patients with relapsed CLL. ashpublications.org The study enrolled sixteen patients who had undergone multiple prior therapies. ashpublications.org While no objective responses were observed according to the 2008 International Workshop on Chronic Lymphocytic Leukemia (IW-CLL) criteria, half of the patients achieved stable disease. ashpublications.org One patient, receiving a 50 mg daily dose, remained on therapy for 12 months. ashpublications.org The trial concluded that this compound is safe for CLL patients and that higher doses may be necessary to achieve a therapeutic effect. ashpublications.org Further research in a Phase II study is planned to assess the efficacy of this compound in treating CLL/SLL (Small Lymphocytic Lymphoma). clinicaltrials.govclinicaltrials.gov
Table 1: Clinical Trial of this compound in Chronic Lymphocytic Leukemia
| Trial Phase | Number of Patients | Key Findings | Reference |
| Phase I | 16 | No objective responses, but 50% of patients achieved stable disease. The drug was found to be safe. | ashpublications.org |
| Phase I/II | Ongoing | To determine the maximum safe dose and efficacy of this compound in relapsed CLL/SLL. | clinicaltrials.govdana-farber.orgcancer.govclinicaltrials.gov |
Liver Cancer
Preclinical studies have suggested that this compound may have a therapeutic role in liver cancer. nih.govnih.gov The rationale for its use in this context is linked to the observation that high levels of activated STAT3 are associated with more aggressive liver tumors. researchgate.net A systematic review of preclinical cancer models identified one study on liver cancer as being well-designed enough to potentially be translated into a clinical trial. nih.govnih.gov This was based on the inclusion of appropriate control and treatment groups, as well as comparison with established chemotherapy drugs. nih.govnih.gov
Esophageal Cancer
Research has also explored the potential of this compound in treating esophageal squamous cell carcinoma (ESCC). aacrjournals.orgresearchgate.net In ESCC, the transcription factor NRF2 is often mutated and overactive, leading to resistance to chemotherapy and radiation. aacrjournals.orgresearchgate.netnih.govaacrjournals.org A small molecule screen identified this compound as an inhibitor of NRF2. aacrjournals.orgresearchgate.netnih.govaacrjournals.org
In preclinical studies using mouse models with a specific NRF2 mutation, oral administration of this compound was shown to suppress the development of esophageal abnormalities associated with high NRF2 activity, without causing any observable toxicity. nih.govaacrjournals.org Further investigation has suggested that this compound acts as a "molecular glue," promoting the binding of NRF2 to its suppressor protein KEAP1, specifically in NRF2-mutated ESCC cells. aacrjournals.orgresearchgate.net This action enhances the sensitivity of cancer cells to chemotherapy and radiation. aacrjournals.orgresearchgate.net These findings have provided the basis for a Phase I clinical trial to investigate this compound in NRF2-mutant ESCC. aacrjournals.orgresearchgate.net A systematic review also concluded that there is potential for this compound to be used in clinical trials for esophageal cancer, particularly in combination with existing chemotherapy drugs. nih.gov
Molecular Pathways Targeted in Cancer Cells
This compound's anticancer effects are attributed to its ability to interfere with specific molecular pathways that are essential for cancer cell function.
DHFR Inhibition in Human Cancer Cells
This compound is a known inhibitor of dihydrofolate reductase (DHFR), an enzyme critical for the synthesis of DNA precursors. nih.govnih.govaacrjournals.orgpatsnap.com By blocking DHFR, this compound disrupts the folate metabolic pathway, leading to a deficiency in reduced folate. nih.govnih.gov This, in turn, inhibits the synthesis of purines and pyrimidines, which are essential building blocks for DNA. nih.gov The effect is particularly pronounced in rapidly dividing cells like cancer cells. patsnap.com
Research has shown that this compound inhibits the activity of human DHFR in various cancer cell lines, including lung cancer cells. aacrjournals.org While both this compound and the well-known DHFR inhibitor methotrexate can inhibit the proliferation of cancer cells by targeting DHFR, this compound has also been observed to inhibit other cancer-promoting processes like epithelial-mesenchymal transition (EMT), metastasis, and invasion, suggesting it may have additional targets. aacrjournals.org The inhibitory effect of this compound on different tumor cell lines has been shown to be positively correlated with the expression level of DHFR. aacrjournals.org
Table 2: Effects of this compound on DHFR and Cancer Cell Proliferation
| Cell Line | Effect of this compound | Reference |
| Lung Cancer Cells (NCI-H460) | Inhibition of DHFR activity | aacrjournals.org |
| Various Cancer Cell Lines | Inhibition of proliferation, positively correlated with DHFR expression | aacrjournals.org |
STAT3 Pathway Inhibition
The Signal Transducer and Activator of Transcription 3 (STAT3) pathway is a key regulator of many processes that promote tumor growth, including inflammation, proliferation, and survival. nih.govnih.gov Persistent activation of STAT3 is a common feature in many human cancers, making it a promising therapeutic target. nih.govnih.govoup.com this compound has been identified as a novel and specific inhibitor of STAT3 transcriptional activity. nih.govnih.gov
Interestingly, this compound does not inhibit the initial steps of STAT3 activation, such as its phosphorylation or movement into the nucleus. nih.gov Instead, it appears to work downstream, inhibiting the ability of STAT3 to activate the transcription of its target genes. nih.gov This inhibitory effect on STAT3 is a consequence of DHFR inhibition and the resulting disruption of folate metabolism. nih.gov The link between folate metabolism and STAT3 activity represents a previously unknown regulatory point in this critical cancer pathway. nih.govnih.gov In preclinical models of breast cancer, this compound has been shown to have both direct tumor-inhibiting and immune-stimulatory effects through its inhibition of STAT3. researchgate.netresearchgate.net
p38-NF-κB Axis Inhibition
In the context of prostate cancer, this compound has been shown to exert its antitumor effects by inhibiting the p38-NF-κB signaling pathway. nih.govresearchgate.netnih.govresearchgate.net Studies on metastatic prostate cancer cell lines (DU145 and PC3) have demonstrated that this compound inhibits cell proliferation, induces cell cycle arrest in the S phase, and promotes apoptosis. nih.govresearchgate.net It also suppressed tumor growth in animal models. nih.govresearchgate.net
The mechanism behind these effects involves the inhibition of the p38 mitogen-activated protein kinase (MAPK). nih.gov Specifically, this compound was found to inhibit the phosphorylation of p38 without affecting the total amount of the p38 protein. nih.gov This inhibition of p38, in turn, affects the nuclear factor kappa B (NF-κB) pathway, a key player in inflammation and cancer progression. nih.gov By suppressing the p38-NF-κB axis, this compound demonstrates a novel mechanism for its antiproliferative and proapoptotic effects in prostate cancer. nih.govresearchgate.netnih.gov
Role of Thymidine Phosphorylase in Cancer
Thymidine phosphorylase (TP), also known as platelet-derived endothelial cell growth factor (PD-ECGF), is an enzyme with a multifaceted and dual role in the landscape of oncology. uah.es It is a key enzyme in the pyrimidine salvage pathway, catalyzing the reversible conversion of thymidine to thymine and 2-deoxy-D-ribose-1-phosphate. nih.govresearchgate.net While this function is primarily catabolic, the significance of TP in cancer extends far beyond its metabolic duties. nih.gov
Upregulation of TP is a common feature in a wide array of solid tumors, including breast, colorectal, gastric, glioblastoma, and lung cancers. uah.esaacrjournals.org This overexpression is often associated with a more aggressive tumor phenotype and a poorer prognosis for patients. uah.esaacrjournals.org The elevated levels of TP in the tumor microenvironment can be triggered by various cellular stressors such as hypoxia, low pH, and exposure to radio- and chemotherapy. uah.esnih.gov
The pro-tumorigenic effects of TP are largely attributed to its angiogenic and anti-apoptotic properties. uah.esnih.gov TP stimulates the growth of new blood vessels, a process crucial for tumor expansion and metastasis, by promoting the migration of endothelial cells. uah.es One of its metabolites, 2-deoxy-D-ribose, acts as a chemoattractant for endothelial cells. nih.gov Furthermore, TP can protect cancer cells from apoptosis induced by various stimuli, including hypoxia and DNA-damaging agents. uah.es
Conversely, TP plays a critical role in the activation of certain chemotherapeutic agents, most notably the 5-fluorouracil (5-FU) prodrug capecitabine. uah.esresearchgate.netingentaconnect.com TP converts capecitabine into its active, cytotoxic form, 5-FU, within the tumor tissue. This has led to therapeutic strategies that aim to induce TP expression to enhance the efficacy of capecitabine-based chemotherapy. uah.es
Recent research has identified TP as a direct target of the antimalarial drug this compound. aacrjournals.orgnih.gov While this compound is known to inhibit dihydrofolate reductase (DHFR), its ability to also target TP contributes to its dual role in inhibiting both cancer cell proliferation and metastasis. aacrjournals.orgnih.gov This finding is particularly relevant as data from The Cancer Genome Atlas (TCGA) database reveals a correlation between TP overexpression and poor prognosis in lung cancer patients. aacrjournals.orgnih.gov
Ubiquitin-Mediated Degradation of AIMP2-DX2
Aminoacyl-tRNA synthetase-interacting multifunctional protein 2 (AIMP2) is recognized as a tumor suppressor. However, a splice variant of this protein, AIMP2-DX2, which lacks exon 2, exhibits oncogenic properties. nih.govmdpi.com High expression levels of AIMP2-DX2 are found in various human cancers, particularly lung cancer, where the ratio of AIMP2-DX2 to the full-length AIMP2 increases with tumor progression. nih.gov This elevated AIMP2-DX2 level is associated with poorer patient survival.
The pro-cancer activity of AIMP2-DX2 stems from its ability to interfere with the tumor-suppressive functions of the wild-type AIMP2. For instance, AIMP2-DX2 can competitively inhibit the binding of AIMP2 to proteins like p53, thereby preventing the AIMP2-mediated stabilization and pro-apoptotic activity of p53.
A key regulatory mechanism for protein levels in cells is the ubiquitin-proteasome system, which tags proteins for degradation. It has been discovered that this compound can induce the ubiquitin-mediated degradation of AIMP2-DX2. nih.govmdpi.com In a study involving lung cancer cell lines, this compound was shown to decrease the levels of AIMP2-DX2 in a dose-dependent manner, without affecting the levels of the full-length AIMP2 protein. nih.govmdpi.com Further experiments confirmed that this reduction in AIMP2-DX2 was due to its ubiquitination and subsequent degradation. nih.govmdpi.com
This targeted degradation of the oncogenic AIMP2-DX2 by this compound has shown significant antitumor effects. In a panel of lung cancer cell lines, this compound most potently suppressed the growth of H460 cells, which have high endogenous levels of AIMP2-DX2. nih.govmdpi.com Moreover, in a mouse xenograft model using H460 cells, the administration of this compound led to a significant reduction in tumor size and weight, an effect comparable to the standard chemotherapy drug taxol. nih.govmdpi.com Analysis of the tumor tissue from these mice revealed decreased levels of AIMP2-DX2, confirming the drug's mechanism of action in vivo. mdpi.com These findings highlight the potential of repurposing this compound for the treatment of cancers with high AIMP2-DX2 expression. nih.govmdpi.com
NRF2 Suppression and its Implications
Nuclear factor erythroid 2-related factor 2 (NRF2) is a transcription factor that plays a central role in the cellular response to oxidative stress. nih.govnih.gov While its protective functions are crucial for normal cell homeostasis, aberrant and persistent activation of NRF2 is a common feature in several cancers, including lung and upper aerodigestive cancers. nih.govnih.gov This hyperactivation of NRF2 can promote tumor initiation and progression, and importantly, confers resistance to various cancer therapies, including chemotherapy, radiation therapy, and immune checkpoint inhibitors. nih.govnih.gov
Given the role of NRF2 in promoting cancer cell survival and therapeutic resistance, there is a significant effort to develop inhibitors of this pathway. nih.gov this compound has been identified as an inhibitor of NRF2. nih.govnih.gov Studies have shown that this compound can suppress both NRF2 mRNA and protein levels. nih.gov
The mechanism by which this compound inhibits NRF2 is linked to its known function as an inhibitor of dihydrofolate reductase (DHFR). nih.govwustl.edu Inhibition of DHFR disrupts one-carbon metabolism, which appears to be essential for maintaining the NRF2 signaling pathway. nih.gov In fact, research has demonstrated that the inactivation of DHFR is a prerequisite for the suppression of NRF2 by this compound. nih.govnih.gov Proteomic analyses have revealed that the effects of this compound on cellular protein expression overlap with those of methotrexate, a classical DHFR inhibitor, and include the suppression of the NRF2 oxidative stress response. nih.govnih.gov
Furthermore, some studies suggest that this compound may also promote the ubiquitination and subsequent degradation of the NRF2 protein, thereby reducing its half-life in cancer cells. researchgate.netbiorxiv.orgaacrjournals.org This dual mechanism of inhibiting NRF2 expression and promoting its degradation makes this compound a compelling candidate for targeting NRF2-driven cancers. The suppression of NRF2 by this compound has been shown to inhibit the growth of NRF2-hyperactive esophageal squamous cell carcinoma cells in preclinical models. researchgate.netaacrjournals.org
Table 1: Effects of this compound on AIMP2-DX2 and NRF2
| Target | Effect of this compound | Mechanism of Action | Implication in Cancer |
|---|---|---|---|
| AIMP2-DX2 | Decreased protein levels | Induces ubiquitin-mediated degradation | Inhibition of tumor growth, particularly in cancers with high AIMP2-DX2 expression like lung cancer. |
| NRF2 | Suppression of mRNA and protein levels | Inhibition of DHFR and one-carbon metabolism; potential promotion of ubiquitination and degradation | Overcoming therapeutic resistance; inhibition of tumor progression in NRF2-driven cancers. |
Combinatorial Therapeutic Strategies with this compound in Oncology
Sensitization to Temozolomide
Temozolomide (TMZ) is a standard-of-care alkylating agent used in the treatment of glioblastoma and metastatic melanoma. nih.govoaepublish.com However, both intrinsic and acquired resistance to TMZ pose significant clinical challenges. oaepublish.com One of the key mechanisms of resistance involves the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT), which removes the cytotoxic lesions induced by TMZ. jbuon.com
Research has shown that this compound can enhance the cytotoxic effects of temozolomide in cancer cells, effectively sensitizing them to the drug. nih.govnih.gov In a screening of 2,000 compounds, this compound was identified as a molecule that, while having little intrinsic cytotoxicity on its own, significantly enhanced the growth-inhibitory effects of TMZ in melanoma cell lines. nih.gov The combination of this compound and TMZ resulted in a synergistic inhibition of cell proliferation. nih.gov
This sensitization is attributed to the antifolate activity of this compound. nih.gov The combination treatment leads to an increase in DNA double-strand breaks and apoptosis, along with cell cycle arrest. nih.gov Notably, the enhanced cell death induced by the combination can be rescued by the administration of leucovorin, a reduced folate, which confirms the role of folate antagonism in this synergistic interaction. nih.gov An important finding is that the sensitizing effect of this compound to TMZ appears to be independent of the MGMT status of the cancer cells, making it a potentially valuable strategy for both MGMT-expressing and MGMT-negative tumors. jbuon.com
The potential of this combination has also been explored in glioblastoma. In vitro studies using glioblastoma stem-like cells and primary glioblastoma cells have demonstrated that this compound enhances the cytotoxic effect of TMZ and sensitizes these cells to radiation therapy. jbuon.comnih.gov Given that this compound is an orally available drug that can cross the blood-brain barrier, its combination with TMZ holds promise for improving the treatment of brain tumors like glioblastoma. nih.gov
Combination with Standard Chemotherapy Drugs
The potential of this compound as an adjunct to standard chemotherapy extends beyond its combination with temozolomide. Its multifaceted mechanisms of action, including the inhibition of DHFR, STAT3, and thymidine phosphorylase, make it a candidate for combination therapies in various cancers. mdpi.comoncoscience.usnih.gov
In the context of glioblastoma, the combination of this compound with the repurposed antifungal drug itraconazole and temozolomide has been investigated. oup.comugent.be In vitro studies using primary glioblastoma patient-derived stem cell lines showed that both itraconazole and this compound, at clinically relevant concentrations, reduced cell viability. oup.comugent.be The combination of these two repurposed drugs with low-dose temozolomide resulted in a significant decrease in cell viability compared to temozolomide monotherapy. oup.comugent.be Synergy analysis indicated mainly additive effects between itraconazole and this compound. oup.com
Another preclinical study investigated the combination of this compound with sorafenib, a multi-kinase inhibitor used in the treatment of various cancers. In a mouse model, the combination of this compound and sorafenib demonstrated a clear synergistic effect in reducing tumor size compared to either drug alone or the control group. nih.gov
The rationale for these combinations often lies in targeting multiple, often complementary, cancer-driving pathways. For instance, in a proposed multidrug adjunctive cancer treatment regimen, this compound is included alongside drugs like celecoxib, disulfiram, and itraconazole to interfere with common growth-driving elements in cancers such as cholangiocarcinoma, colon adenocarcinoma, and non-small-cell lung cancer. mdpi.com
Table 2: Preclinical Combinatorial Studies with this compound
| Combination Drug | Cancer Model | Key Findings |
|---|---|---|
| Temozolomide | Melanoma, Glioblastoma | Synergistic inhibition of cell proliferation; increased DNA damage and apoptosis; sensitization to TMZ is independent of MGMT status. |
| Itraconazole and Temozolomide | Glioblastoma | Additive effects between this compound and itraconazole; significant decrease in cell viability with the triple combination compared to TMZ alone. |
| Sorafenib | Mouse cancer model | Synergistic reduction in tumor size. |
Pharmacokinetic and Pharmacodynamic Research
Population Pharmacokinetics of Pyrimethamine
Population pharmacokinetic (PK) modeling, utilizing a nonlinear mixed-effects approach, has been instrumental in understanding the behavior of this compound in various patient groups. nih.govbps.ac.uk These models typically describe the concentration-time data of this compound using a one-compartment open model with first-order absorption and elimination, particularly when rich data in the initial hours after administration are not available. nih.gov However, two-compartment models have also been employed, especially in studies with more intensive sampling. asm.org
Children: The pharmacokinetics of this compound in children, particularly those with congenital toxoplasmosis, show significant variability. nih.gov Studies have utilized population PK models to characterize drug disposition in this population, ranging from newborns to adolescents. nih.govbps.ac.uk In children from 1 week to 14 years old, a one-compartment open model has been shown to best describe the plasma concentration-time profiles of this compound. bps.ac.uk For children treated for congenital toxoplasmosis, a nonparametric modeling analysis also supported a one-compartment model. nih.gov The pharmacokinetic parameters are notably influenced by developmental changes, with body weight being a significant covariate. nih.govbps.ac.uk
Pregnant Women: Pregnancy induces significant physiological changes that alter the pharmacokinetics of this compound. nih.govnih.gov Population pharmacokinetic analyses have revealed that pregnancy increases both the clearance and the volume of distribution of this compound. asm.orgresearchgate.net One study involving pregnant women in their second or third trimester found that clearance was 17.5% lower, while another reported an increased relative clearance of 48%. nih.govasm.org These discrepancies may be attributed to differences in study design, such as the use of a nonpregnant comparator group versus an early postpartum follow-up. nih.gov The volume of distribution for this compound has also been shown to increase during pregnancy. asm.orgasm.org
Several patient-specific factors, or covariates, have been identified as having a significant impact on the pharmacokinetics of this compound.
Body Weight: This is a consistently significant covariate in pediatric populations. nih.govbps.ac.uk Allometric scaling of pharmacokinetic parameters with total body weight has been shown to improve model fit. smc-alliance.orgasm.org Clearance and volume of distribution are significantly related to body weight. bps.ac.uk
Age: Age has also been identified as a significant covariate, particularly in children, where it influences clearance maturation. smc-alliance.orgasm.org One study noted that in children with congenital toxoplasmosis, age significantly influenced this compound pharmacokinetics. nih.gov
Nutritional Status: The nutritional status of a patient, often measured by the weight-for-age Z-score, can affect this compound's bioavailability. smc-alliance.orgasm.org Underweight-for-age children have been found to have a 26.7% lower bioavailability of this compound for each Z-score unit below -2. smc-alliance.orgasm.org
Pregnancy: As a covariate, pregnancy significantly alters this compound's pharmacokinetic parameters. Studies have shown an increase in the relative clearance by 48% and an increase in the relative volumes of distribution of the central and peripheral compartments by 46% and 99%, respectively. asm.org Another study reported an 18% decrease in clearance during pregnancy. nih.govnih.govresearchgate.net
Table 1: Influence of Covariates on this compound Pharmacokinetic Parameters
| Covariate | Effect on Pharmacokinetics | Patient Population | Source |
|---|---|---|---|
| Body Weight | Significantly influences clearance and volume of distribution. | Children | nih.govbps.ac.uk |
| Age | Significantly influences clearance maturation. | Children | smc-alliance.orgasm.org |
| Nutritional Status | Underweight status is associated with lower bioavailability. | Children | smc-alliance.orgasm.org |
| Pregnancy | Increases relative clearance and volume of distribution. | Pregnant Women | asm.org |
| Decreases clearance. | Pregnant Women | nih.govnih.govresearchgate.net |
The co-administration of other drugs can alter the pharmacokinetics of this compound. A notable example is the interaction with azithromycin. When this compound is given with azithromycin, its clearance is significantly increased. nih.gov One study found that while pregnancy alone increased the relative clearance of this compound by 48%, co-administration with azithromycin resulted in a greater increase of 80%. asm.org This suggests a clinically significant interaction that may require dose adjustments. nih.gov Limited data are available on interactions with other drugs. hep-druginteractions.org
There is a substantial difference in the plasma half-life of this compound between humans and mice, which is a critical consideration when translating preclinical research to human clinical trials. nih.govoup.com
Humans: The plasma half-life of this compound in humans is approximately 96 hours. nih.govoup.comfda.govdrugbank.com
Mice: In mice, the half-life is significantly shorter, around 5 to 6 hours. nih.govoup.com
This marked difference in elimination rates means that direct translation of dosages from mouse models to humans based on body weight alone is not appropriate and requires careful pharmacokinetic consideration. nih.govoup.com
Table 2: Comparison of this compound Half-life in Humans and Mice
| Organism | Approximate Half-life | Source |
|---|---|---|
| Human | 96 hours | nih.govoup.comfda.govdrugbank.com |
| Mouse | 5-6 hours | nih.govoup.com |
Drug Interactions Affecting this compound Pharmacokinetics (e.g., co-administration with Azithromycin)
Pharmacodynamic Endpoints and PK/PD Modeling
Pharmacokinetic/pharmacodynamic (PK/PD) modeling aims to establish a relationship between drug exposure (pharmacokinetics) and the pharmacological effect (pharmacodynamics). nih.govplos.org For this compound, this involves linking its concentration in the body to its therapeutic efficacy against parasites like Toxoplasma gondii and Plasmodium falciparum. drugbank.comiddo.org
The therapeutic effect of this compound is dependent on maintaining adequate drug concentrations over time. The highly synergistic relationship between this compound and sulfadoxine has made defining specific therapeutic concentrations challenging. iddo.org However, studies have shown that suboptimal drug exposure can lead to treatment failure and the development of resistance. smc-alliance.org For instance, in the context of toxoplasmic encephalitis in HIV-infected patients, a 1-year incidence of the disease was lower in patients receiving this compound compared to a placebo, highlighting a dose-response relationship. oup.com Similarly, a high rate of reemergent parasitemia in pregnant women treated for malaria may indicate a suboptimal response to standard doses, suggesting that the lower plasma concentrations observed during pregnancy could compromise efficacy. asm.org PK/PD modeling is a crucial tool to refine dosing regimens to ensure adequate drug exposure and therapeutic success in different patient populations. iddo.org
Correlation between Pharmacokinetic Parameters and Clinical Outcomes (e.g., treatment failure, resistance development)
The clinical efficacy of this compound is intrinsically linked to its pharmacokinetic (PK) profile. Research has established a significant correlation between specific PK parameters, such as plasma drug concentration and elimination half-life, and clinical outcomes, including treatment success, failure, and the emergence of drug-resistant parasite strains. The interplay between drug exposure and parasite susceptibility is a critical determinant in the therapeutic success of this compound-containing regimens.
Detailed Research Findings
Studies investigating the use of sulfadoxine-pyrimethamine (SP) for treating Plasmodium falciparum malaria have provided direct evidence linking drug concentrations to clinical results. A key factor in treatment failure is the presence of parasites with genetic mutations that confer resistance. researchgate.netasm.org However, even in the face of resistant genotypes, adequate drug exposure can play a decisive role in clearing the infection.
Conversely, suboptimal drug concentrations are a risk factor for treatment failure. researchgate.netresearchgate.net In one multivariate analysis, a low day 3 blood concentration of this compound was identified as a risk factor for late treatment failure. nih.gov Physiological conditions that alter pharmacokinetics can also impact outcomes. For instance, pregnancy is associated with a significantly lower area under the concentration-time curve (AUC) for this compound, which could compromise both curative efficacy and the prophylactic effect of the drug. asm.org
The long elimination half-life of this compound is another critical pharmacokinetic parameter that influences the development of resistance. asm.orgnih.govnih.gov While beneficial for maintaining therapeutic concentrations, a long half-life creates a prolonged period where sub-therapeutic drug levels persist in the body. This extended window provides a strong selective pressure for existing parasites with resistance-conferring mutations, allowing them to survive and propagate. asm.orgnih.govnih.gov Modeling studies have shown that this selection is the first stage in the development of widespread clinical resistance. asm.orgnih.gov
However, the relationship is not always direct. A study in Tanzania found uniform efficacy of SP even with reduced doses, but this was in a context where all tested parasite isolates showed full sensitivity in vitro. oup.com In another case, treatment failure in an individual was not attributable to parasite resistance or inadequate plasma drug levels, but rather to an undefined host factor, demonstrating the complexity of the host-parasite-drug interaction. sci-hub.se
Interactive Data Table: this compound Concentration and Clinical Outcome in Malaria Treatment
The following table summarizes findings from studies correlating this compound plasma concentrations with treatment outcomes.
| Study Population & (Parasite) | Parameter Measured | Concentration in Patients with Successful Outcome (ACPR) | Concentration in Patients with Treatment Failure (TF) | Key Finding |
| Children in Gabon ird.fr | Day 3 Plasma this compound | >175 ng/ml | <175 ng/ml | A concentration of 175 ng/ml was a significant breakpoint for predicting treatment success (Odds Ratio: 11.8). ird.fr |
| Children in Malawi researchgate.netasm.org | Day 3 Blood this compound (Overall) | 205 ng/ml (mean) | 172 ng/ml (mean) | The difference in the overall population was not statistically significant (P=0.25). researchgate.netasm.org |
| Children in Malawi with "Quintuple Mutant" P. falciparum researchgate.netasm.org | Day 3 Blood this compound | 305 ng/ml (mean) | 228 ng/ml (mean) | Higher this compound concentration was significantly associated with clearing a resistant infection (P=0.037). researchgate.netasm.org |
Advanced Synthetic Methodologies and Analog Development
Synthetic Routes for Pyrimethamine
The traditional synthesis of this compound, chemically known as 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidine, typically commences with p-chlorophenylacetonitrile. wikipedia.orgwikimedia.org This starting material undergoes a condensation reaction with an ethyl propionate ester in the presence of a strong base, such as sodium methoxide, to yield a β-ketonitrile intermediate. wikimedia.orgchemicalbook.com This intermediate exists in tautomeric equilibrium with a more stable enol form. wikimedia.org
The subsequent step involves the methylation of the enol to form an enol ether. wikimedia.org Historically, this has been achieved using diazomethane, a reagent that is both toxic and explosive. wikimedia.org A less hazardous alternative involves the use of orthoformates, such as ethyl orthoformate, to produce a methoxymethylene derivative. wikimedia.orgchemicalbook.com
The final stage of the synthesis is the condensation of the enol ether with guanidine in refluxing ethanol to form the pyrimidine ring of this compound. wikimedia.org Since guanidine is a strong base, it is typically prepared in situ from a salt like guanidine hydrochloride and a base such as sodium ethoxide. wikimedia.org This reaction mixture is sensitive to air, as atmospheric carbon dioxide can readily form guanidinium carbonate. wikimedia.org
A patented method for synthesizing the intermediate p-chlorobenzonitrile involves the reaction of p-chlorobenzyl with stannous chloride and cyanamide. google.comgoogle.com The process includes heating the mixture, followed by cooling, separation, and purification steps including washing, vacuum distillation, and recrystallization to obtain the desired intermediate. google.comgoogle.com
Design and Synthesis of Novel this compound Analogues
The development of novel this compound analogues is a critical area of research aimed at overcoming drug resistance and improving therapeutic efficacy.
To enhance the potency and selectivity of this compound, researchers have focused on modifying its structure to exploit differences between the target enzyme in pathogens, dihydrofolate reductase (DHFR), and the human form of the enzyme (hDHFR). For instance, in the context of Toxoplasma gondii (TgDHFR), a meta-biphenyl analog was designed that improved both selectivity and potency compared to the parent compound. acs.org
Further optimization led to the discovery of an arylpiperazine series of compounds. acs.org One standout, a 5-pyrimidine analog, demonstrated a 307-fold selectivity for TgDHFR over hDHFR and a significantly improved solubility profile. acs.org The introduction of electron-donating groups, such as a methyl or methoxy group, on the pyrimidine ring was predicted to enhance potency and selectivity by favorably interacting with the pathogen's enzyme while having an unfavorable interaction with the human enzyme. acs.org Specifically, a methyl analog maintained high potency for TgDHFR with a 237-fold selectivity. acs.org Another potent derivative, WCDD115, was developed and showed a 31-fold greater potency in inhibiting hDHFR compared to this compound. biorxiv.orgnih.gov
A primary mechanism of resistance to this compound in malaria parasites like Plasmodium falciparum and Plasmodium vivax involves point mutations in the DHFR enzyme. researchgate.netpnas.org A common mutation is S108N in P. falciparum and the corresponding S117N in P. vivax, which creates steric hindrance with the p-chlorophenyl group of this compound, reducing its binding affinity. pnas.orgasm.org
To overcome this, analogues have been designed with modifications to the 5-phenyl ring to avoid this steric clash. researchgate.net Meta-substituted analogues, such as the meta-chloro and meta-bromo derivatives, have shown significantly improved inhibitory activity against both wild-type and mutant DHFR enzymes, including highly resistant quadruple-mutant strains. researchgate.net For example, the meta-bromo analog was a much more powerful inhibitor of the quadruple-mutant DHFR than this compound itself. researchgate.net Another approach involves designing analogues that only occupy the substrate space of the active site, thereby retaining their affinity for mutant enzymes. pnas.org An analog lacking the p-chloro group, for instance, was shown to effectively inhibit both wild-type and a double-mutant P. vivax DHFR. pnas.org
Structure-activity relationship (SAR) studies are crucial for understanding how different chemical modifications affect the biological activity of this compound analogues. For instance, a series of 25 this compound-based derivatives were synthesized to explore the SAR for the inhibition of the NRF2 pathway. biorxiv.org Modifications were made to the 4-amino moiety, the 6-ethyl moiety, and the chlorine on the 5-para-chlorobenzene ring. biorxiv.org This study led to the identification of WCDD115, which was 22-fold more potent than this compound in inhibiting NRF2. biorxiv.orgnih.gov
In the context of antimalarial activity, SAR studies have shown that modifications at the 5-phenyl ring of the 2,4-diaminopyrimidine core can significantly improve activity against resistant parasites. tandfonline.com Three-dimensional quantitative structure-activity relationship (3D-QSAR) studies, such as comparative molecular field analysis (CoMFA), have been employed to investigate the steric and electrostatic fields of this compound derivatives to understand their interaction with the target enzyme. tandfonline.com These studies help in the rational design of more potent inhibitors.
To combat the development of drug resistance, researchers are exploring the development of derivatives that can inhibit multiple targets. In Trypanosoma brucei, the causative agent of African trypanosomiasis, this compound has been shown to be a dual inhibitor of both dihydrofolate reductase (DHFR) and pteridine reductase 1 (PTR1), two key enzymes in the folate biosynthesis pathway. mdpi.com This dual-targeting ability is a significant finding, as PTR1 can compensate for DHFR inhibition, leading to drug resistance. mdpi.com
Furthermore, hybrid inhibitors have been designed that combine the structural features of both rigid (like this compound) and flexible inhibitors in a single molecule. acs.org These hybrids are effective against both wild-type and multiple mutant strains of P. falciparum DHFR. acs.org For example, a hybrid compound, BT1, showed very high affinities for both wild-type and mutant P. falciparum DHFRs with high selectivity over the human enzyme. acs.org This approach aims to forestall the emergence of resistance by targeting the enzyme in multiple ways. acs.org
Exploration of Structure-Activity Relationships (SAR)
Green Chemistry Approaches in this compound Synthesis
Traditional methods for synthesizing pyrimidines, including this compound, often rely on hazardous solvents and toxic reagents. rasayanjournal.co.in In recent years, there has been a growing emphasis on developing more environmentally friendly and sustainable "green chemistry" approaches. These methods aim to reduce waste, shorten reaction times, and use safer substances. rasayanjournal.co.injocpr.com
For the synthesis of pyrimidine derivatives in general, several green techniques have been explored, such as microwave-assisted synthesis, solventless reactions, and the use of mechanochemistry. rasayanjournal.co.innih.gov Mechanochemical synthesis, which uses mechanical energy to initiate reactions, can be more efficient and sustainable than traditional solvent-based methods. nih.govresearchgate.net This approach aligns with the green chemistry principles of energy efficiency and waste reduction. nih.gov A mechanochemical protocol for the synthesis of this compound has been described as an easy and efficient method. nih.gov These green approaches not only offer environmental benefits but can also lead to higher yields and simpler purification processes. rasayanjournal.co.in
Future Research Directions and Translational Perspectives
Novel Therapeutic Applications of Pyrimethamine
Beyond its traditional use, the biological activities of this compound are being reassessed, revealing potential applications in a broader range of diseases. Researchers are actively repurposing this dihydrofolate reductase (DHFR) inhibitor for emerging viral infections and exploring its mechanisms of action beyond its effects on folate metabolism.
The established mechanism of this compound, which involves the inhibition of DHFR and subsequent disruption of nucleic acid synthesis, has prompted investigations into its efficacy against various pathogens. nih.gov High-throughput screening of existing drug libraries has identified this compound as a candidate for treating several emerging viral diseases.
Zika Virus (ZIKV): this compound has been identified as an inhibitor of Zika virus infection. nih.govasm.org As a folic acid antagonist, it interferes with DNA and RNA synthesis, a critical process for viral replication. nih.gov Currently, there is no specific antiviral therapy available for ZIKV, making the repurposing of approved drugs like this compound a significant area of research. nih.govcda.gov.sgmdpi.com
Dengue Virus (DENV): Through computational screening, this compound was found to have a chemical structure similar to known inhibitors of the dengue NS2B/3 proteinase. asm.org Subsequent studies showed it could reduce dengue virus titers with a 50% inhibitory concentration (IC50) of 1.2 μM and lower the viral burden in the spleens of infected mice. asm.org The proposed mechanism involves the inhibition of intramolecular NS2B/3 cleavage. asm.org
Rabies Virus (RABV): In vitro studies have demonstrated that this compound can inhibit rabies virus replication. dovepress.comresearchgate.net The mechanism is believed to be the inhibition of adenosine synthesis. researchgate.net However, this inhibitory effect observed in cell cultures did not translate to efficacy in infected mice, potentially because the drug interfered with the innate immune response. dovepress.comresearchgate.net
Human Immunodeficiency Virus (HIV): The effect of this compound on HIV-1 replication is complex. While some research indicates it can enhance HIV-1 replication by causing an accumulation of cells in the S-phase, it has also been shown to increase the antiviral activity of certain antiretroviral drugs like zidovudine and stavudine by approximately four-fold. dovepress.comnih.gov
Table 1: Investigational Antiviral Activities of this compound
| Virus | Finding | Proposed Mechanism of Action | Reference |
| Zika Virus (ZIKV) | Inhibits ZIKV infection in vitro and in mouse models. | Inhibition of dihydrofolate reductase (DHFR), blocking DNA/RNA synthesis. | nih.govasm.org |
| Dengue Virus (DENV) | Reduces viral titers in vitro and viral load in mice. | Inhibition of viral NS2B/3 proteinase cleavage. | asm.org |
| Rabies Virus (RABV) | Inhibits viral replication in vitro. | Inhibition of adenosine synthesis. | researchgate.net |
| HIV-1 | Potentiates the activity of antiretrovirals like zidovudine and stavudine. | Affects the cellular machinery, leading to S-phase accumulation. | dovepress.comnih.gov |
Recent research has uncovered biological activities of this compound that extend beyond its established roles, opening doors to potential treatments for genetic and inflammatory disorders.
Neurological Disorders: this compound is being investigated as a pharmacological chaperone for treating Late-Onset Tay-Sachs disease (LOTS), a neurodegenerative lysosomal storage disorder caused by mutations in the gene for the α-subunit of the β-hexosaminidase A (HexA) enzyme. researchgate.net In vitro studies on patient fibroblasts have shown that this compound can act as a chaperone for HexA, increasing its intracellular levels and activity. researchgate.netclinicaltrials.govclinicaltrials.gov This suggests it could help restore the enzyme's function in breaking down GM2-ganglioside, the accumulation of which leads to neurodegeneration. clinicaltrials.govclinicaltrials.gov
STAT3 Pathway Inhibition: this compound has been identified as a novel and specific inhibitor of the transcriptional activity of Signal Transducer and Activator of Transcription 3 (STAT3). researchgate.net STAT3 is a key regulator in many pro-tumorigenic processes. researchgate.net The inhibitory effect of this compound on STAT3 is a downstream consequence of its primary action on human DHFR, which leads to a deficiency in reduced folate and subsequently impacts STAT3's ability to regulate gene expression. researchgate.netnih.gov This discovery reveals a previously unknown link between folate metabolism and the STAT3 signaling pathway. nih.gov
Repurposing this compound for Emerging Disease Treatment
Overcoming Drug Resistance Challenges
The clinical utility of this compound, particularly in treating malaria, has been significantly hampered by the emergence and spread of drug-resistant parasites. tandfonline.comnih.gov Resistance primarily arises from specific point mutations in the parasite's dhfr gene, which reduce the drug's binding affinity to the enzyme. cambridge.orgwalshmedicalmedia.com Future research is intensely focused on strategies to circumvent these resistance mechanisms.
A key strategy to combat this compound resistance is the rational design of new DHFR inhibitors that are effective against mutant forms of the enzyme. cambridge.orgbiorxiv.org
Structure-Based Drug Design: Crystal structures of P. falciparum DHFR, both wild-type and mutant, have provided a molecular basis for understanding resistance. cambridge.org The S108N mutation, a key driver of this compound resistance, creates steric hindrance that blocks the binding of rigid inhibitors like this compound. researchgate.netacs.org Researchers are developing inhibitors with flexible side chains that can adapt to the altered shape of the mutant enzyme's active site, thereby avoiding this steric clash and retaining efficacy. cambridge.orgresearchgate.net
Novel Antifolates: P218 is a promising next-generation DHFR inhibitor designed specifically to be effective against this compound-resistant malaria strains. biorxiv.orgmdpi.com It is a selective inhibitor of the parasite's DHFR and has shown activity against parasites with the quadruple mutant dhfr gene (N51I+C59R+S108N+I164L), which confers complete resistance to this compound. biorxiv.orgmdpi.com P218 was designed with a flexible side-chain and a carboxylate group to form strong bonds with conserved residues in the enzyme's active site that are not affected by common resistance mutations. nih.gov Other novel antifolates, such as the dihydrotriazine WR99210, also show effectiveness against resistant parasites due to their structural flexibility. cambridge.orgbiorxiv.org
Table 2: Comparison of DHFR Inhibitors
| Compound | Generation/Class | Key Feature | Efficacy Against Resistant Strains | Reference |
| This compound | First-Generation Antifolate | Rigid chemical structure. | Low efficacy against strains with S108N and other mutations. | cambridge.orgacs.orgoup.com |
| P218 | Next-Generation Antifolate | Flexible side-chain designed to bind conserved residues. | Highly potent against this compound-resistant strains, including quadruple mutants. | biorxiv.orgmdpi.comnih.gov |
| WR99210 | Dihydrotriazine Derivative | Flexible side-chain. | Effective against this compound-resistant parasites. | cambridge.orgbiorxiv.org |
| Chlorcycloguanil | Biguanide | Short-acting antifolate. | Retains activity against some this compound/sulfadoxine-resistant parasites. | oup.com |
In addition to developing new drugs, strategies are being implemented to slow the development and spread of resistance to existing and new antifolates.
Combination Therapies: Combining this compound with other antimalarial agents, such as sulfadoxine (in sulfadoxine-pyrimethamine, SP) or artemisinin derivatives (in Artemisinin-based Combination Therapies, ACTs), is a cornerstone of antimalarial strategy. nih.govwalshmedicalmedia.comwho.int This approach reduces the likelihood of resistant parasites emerging and enhances treatment efficacy by targeting different metabolic pathways simultaneously. nih.govwalshmedicalmedia.com For example, combining an antifolate with an artemisinin derivative can help clear parasites rapidly and reduce the transmission of resistant strains. nih.govnih.gov
Alternative Drug Combinations: As resistance to SP has become widespread, research into alternative combinations is critical. tandfonline.comwho.int The combination of amodiaquine and SP has shown high efficacy in some regions. plos.org The development of combinations using short-acting antifolates like chlorproguanil with dapsone was pursued to exert less selective pressure for resistance compared to the long-acting this compound. oup.com
Drug Cycling and Adaptive Therapy: More dynamic approaches, such as periodically alternating between different drug regimens (drug cycling) or adjusting treatments based on real-time monitoring of parasite susceptibility (adaptive therapy), are being considered. walshmedicalmedia.com These strategies aim to manage the evolutionary pressure that drives resistance, potentially prolonging the therapeutic lifespan of antimalarial drugs. walshmedicalmedia.comoup.com
Development of Next-Generation DHFR Inhibitors
Integration of Omics Technologies in this compound Research
The application of high-throughput "omics" technologies—genomics, proteomics, and metabolomics—is revolutionizing this compound research. These tools provide a system-wide view of the drug's effects and the complex mechanisms of resistance.
Genomics: Population genomic studies have been crucial for mapping the genes associated with this compound resistance. researchgate.net By sequencing the genomes of P. falciparum isolates from different regions, researchers can track the prevalence and spread of resistance-conferring mutations in the pfdhfr and pfdhps genes. asm.org This surveillance is essential for informing treatment policies and identifying areas where SP-based therapies are likely to fail. asm.orgnih.gov For instance, genomic analysis can confirm the near-fixation of triple-mutant pfdhfr haplotypes (N51I, C59R, S108N) in certain parasite populations, indicating high levels of this compound resistance. asm.org
Proteomics and Metabolomics: Quantitative proteome profiling and metabolomic analyses are being used to identify the direct molecular targets of this compound and elucidate its broader mechanism of action. researchgate.netnih.gov For example, a proteome-wide approach identified human DHFR as the target through which this compound inhibits STAT3 transcriptional activity. nih.gov This was coupled with metabolomic analysis, which confirmed that the drug's effect was due to a deficiency in reduced folates. researchgate.net Such studies can uncover unexpected regulatory nodes and provide deeper insights into the drug's impact on cellular metabolism, both in the parasite and the host. nih.govplos.orgnih.govmdpi.com
Genomics and Proteomics for Target Identification and Mechanism Elucidation
The advent of "omics" technologies has significantly advanced our understanding of this compound's mechanisms of action beyond its well-established role as a dihydrofolate reductase (DHFR) inhibitor. aacrjournals.org Genomic and proteomic approaches are pivotal in identifying novel drug targets and elucidating the complex cellular responses to this compound treatment.
Chemoproteomics, a key technology in this field, allows for the direct and unbiased identification of protein targets of small molecules like this compound. nih.gov This is particularly valuable for drugs discovered through phenotypic screening, where the molecular target may not be immediately known. nih.gov Techniques such as affinity-based target identification and activity-based protein profiling (ABPP) are instrumental in this process. thno.org For instance, proteomics analysis of lung cancer cells treated with this compound revealed thymidine phosphorylase (TP) as a target protein, in addition to human DHFR (hDHFR). aacrjournals.org This dual-targeting capability suggests a broader anti-cancer role for this compound, impacting both proliferation and metastasis. aacrjournals.org
In the context of infectious diseases, chemo-proteomic methods have been employed to identify targets in parasites like Plasmodium falciparum. nih.gov These approaches can uncover poly-pharmacology, a common feature of effective drugs, and identify off-target binding that could have implications for human toxicity. nih.gov Furthermore, proteomic studies on Toxoplasma gondii have shown that this compound treatment leads to altered expression levels of numerous newly synthesized proteins involved in protein synthesis, stress responses, and energy metabolism, indicating complex adaptive responses by the parasite. mdpi.com
The integration of these "omics" approaches provides a more comprehensive picture of this compound's interactions within the cell, paving the way for the development of more effective therapeutic strategies and the identification of potential biomarkers for drug response.
Metabolomics to Understand Metabolic Pathway Perturbations
Metabolomics, the large-scale study of small molecules within cells, has emerged as a powerful tool to understand the metabolic perturbations induced by this compound. nih.gov By analyzing the global metabolic changes in response to the drug, researchers can gain insights into its mechanism of action and identify affected biochemical pathways. nih.govresearchgate.net
In the study of antimalarial drugs, widely targeted and untargeted metabolomics approaches have revealed unique metabolic phenotypes for several compounds, including this compound. nih.gov These studies have demonstrated that the metabolic disruptions caused by antimalarial compounds are directly related to their specific modes of action. nih.gov For example, metabolomic profiling of Plasmodium falciparum treated with this compound has confirmed its role in targeting folate biosynthesis, as evidenced by perturbations in metabolites like dUMP. asm.org
This approach is not limited to malaria research. Metabolomics has been used to investigate the effects of this compound in combination with other drugs, such as sulfadiazine, on Toxoplasma gondii infections, revealing the biochemical pathways associated with the treatment. researchgate.net Furthermore, metabolomics databases, such as the Metabolomics Workbench, serve as repositories for data from studies on this compound and other compounds, facilitating broader analysis and integration of findings. metabolomicsworkbench.org
The comprehensive data generated through metabolomics can help in classifying compounds based on their metabolic signatures, triaging new lead compounds, and defining their specific modes of action, which is crucial for the development and optimization of new therapeutic agents. nih.gov
Advanced Preclinical Models and Translational Studies
Refinement of in vitro and in vivo Models for Efficacy and Toxicity Assessment
The effective translation of this compound from preclinical discovery to clinical application heavily relies on the use of robust and refined in vitro and in vivo models. These models are essential for assessing both the efficacy and potential toxicity of the drug.
In vitro models are fundamental for initial screening and mechanistic studies. For instance, the in vitro efficacy of this compound has been evaluated against various parasites, including Babesia duncani and Toxoplasma gondii. biorxiv.orgscielo.br Studies have determined the half-maximal inhibitory concentration (IC50) of this compound against different parasite strains, providing a quantitative measure of its potency. scielo.br For example, against T. gondii tachyzoites, this compound was effective at an IC50 of 0.482µM. scielo.br The development of novel in vitro culture systems that allow for the comparison of drug efficacy across different parasite species and isolates is a significant refinement. biorxiv.org
In vivo models, primarily in animals, are critical for evaluating the systemic effects of this compound. Mouse models of various cancers have been instrumental in demonstrating the tumor-suppressing capabilities of this compound. oup.comnih.gov These studies have investigated different routes of administration and dosages to assess efficacy. nih.gov However, a systematic review of these preclinical cancer models highlighted that many studies had a moderate risk of bias and did not always adhere to transparent reporting guidelines like the ARRIVE guideline. nih.gov This underscores the need for more rigorously designed in vivo studies. Furthermore, the choice of animal model and cell lines is crucial; for example, some studies used mouse-derived cancer cell lines in immunocompetent mice, which may not fully recapitulate the human tumor microenvironment. nih.gov
Refinements in these models, such as the use of humanized mouse models and the careful selection of cell lines, are necessary to improve the predictive value of preclinical studies.
Bridging Preclinical Findings to Human Clinical Trials
The transition of this compound from promising preclinical results to successful human clinical trials is a complex process that requires careful consideration of pharmacokinetic and pharmacodynamic differences between species. oup.com A major challenge is the direct translation of effective doses from animal models to humans. nih.gov
Pharmacokinetic profiles, particularly the drug's half-life, can differ significantly between species. For instance, the half-life of this compound is approximately 96 hours in humans but only 6 hours in mice. oup.comnih.gov This disparity means that a direct conversion of dosage based on body weight or surface area may not be appropriate and could lead to ineffective or toxic doses in humans. oup.comoup.com Therefore, a more nuanced approach that considers these pharmacokinetic differences is essential for establishing safe and effective dosing regimens in clinical trials. nih.gov
Systematic reviews of preclinical studies play a crucial role in identifying which research shows the highest potential for successful clinical translation. oup.com These reviews assess the quality of the preclinical evidence, including the use of appropriate controls, comparison with standard-of-care drugs, and the relevance of the animal model to the human disease. oup.com For example, a review of this compound in preclinical cancer models suggested that a study on liver cancer showed strong potential for moving into a clinical trial. nih.gov
Q & A
Q. How does pyrimethamine inhibit dihydrofolate reductase (DHFR), and what experimental models are used to validate its efficacy?
this compound competitively inhibits DHFR by binding to its active site, disrupting folate metabolism and nucleotide synthesis. Experimental validation typically involves:
- Enzyme inhibition assays : Measuring IC50 values using recombinant DHFR enzymes (e.g., from Plasmodium falciparum) under varying this compound concentrations .
- Cell-based assays : Assessing parasite growth inhibition in P. falciparum cultures or Escherichia coli models with DHFR complementation systems .
- Flow cytometry : Quantifying cell cycle arrest in cancer models (e.g., colorectal cancer cells) to link DHFR inhibition to antiproliferative effects .
Q. What pharmacokinetic properties of this compound are critical for optimizing dosing regimens in preclinical studies?
Key properties include:
- Bioavailability : this compound’s weak base nature (pKa ~7) limits solubility at gastric pH, requiring formulation adjustments (e.g., solid dispersions) to enhance dissolution .
- Cerebrospinal fluid (CSF) penetration : this compound achieves 25–50% of plasma concentrations in CSF, critical for treating toxoplasmosis .
- Metabolic stability : Hepatic CYP450 metabolism necessitates monitoring for drug-drug interactions in combination therapies . Pharmacokinetic studies use HPLC or gas chromatography (e.g., Hewlett-Packard systems) with electron-capture detection for precision .
Q. How are this compound-resistant mutants generated in laboratory settings, and what genetic markers are monitored?
Resistance is induced via:
- Stepwise drug pressure : Serial passage of parasites (e.g., Plasmodium or Neospora caninum) in increasing this compound concentrations, followed by cloning via limiting dilution .
- Site-directed mutagenesis : Introducing mutations (e.g., S108N, C59R, N51I, I164L) in P. falciparum DHFR to replicate clinical resistance . Resistance is confirmed using allele-specific PCR or sequencing to detect mutations linked to reduced drug binding .
Advanced Research Questions
Q. What evolutionary pathways explain the emergence of high-level this compound resistance in malaria parasites?
Resistance arises through sequential mutations in dhfr:
- Primary mutation : S108N confers low-level resistance (IC50 increases 10–100×).
- Secondary mutations : C59R and N51I amplify resistance (IC50 >1,000×).
- Tertiary mutation : I164L (common in Southeast Asia) elevates IC50 >10,000× but is rare in Africa due to fitness costs . Computational models (e.g., fixation probability based on mutational spectra) predict dominant pathways (e.g., S108N → C59R → N51I → I164L) .
Q. How does this compound induce senescence in colorectal cancer (CRC) cells, and what molecular pathways are involved?
this compound triggers S-phase arrest and senescence via:
- p38MAPK-p53 axis activation : Phosphorylation of p53 upregulates p21, halting cell cycle progression .
- Senescence-associated β-galactosidase (SA-β-gal) : Used as a biomarker in staining assays .
- RNA-seq and Western blotting : Confirm downregulation of cyclins (e.g., Cyclin A2) and upregulation of senescence markers .
Q. What synergies exist between this compound and sulfonamides, and how are these interactions quantified?
Synergistic effects are evaluated via:
- Isobologram analysis : Calculating combination indices (CI <1 indicates synergy) .
- Lesion reduction assays : In Neospora caninum models, combining this compound with sulfadiazine reduces cyst production by >90% . Mechanistically, sulfonamides inhibit dihydropteroate synthase (DHPS), complementing DHFR inhibition .
Q. How can genetic surveillance data inform strategies to mitigate this compound resistance in endemic regions?
- Allele frequency monitoring : Tracking mutations (e.g., S108N, C59R) in dhfr across geographic regions to detect emerging resistance .
- Fitness cost assessments : Mutations like I164L reduce parasite viability in low-drug environments, guiding cyclical drug-use policies .
- Cross-resistance profiling : Testing resistance to cycloguanil or chlorproguanil to identify collateral sensitivity .
Methodological Guidance
Q. What statistical approaches are optimal for analyzing this compound resistance data in parasite cultures?
- Mann-Whitney U test : Compare lesion counts or growth rates between drug-treated and control groups .
- Linear regression : Model dose-response curves for IC50 determination in enzyme inhibition assays .
- Fixation probability models : Simulate mutation trajectories using empirical mutational spectra .
Q. How can researchers address contradictions in this compound’s apoptotic vs. senescent effects across cancer models?
Q. What ethical and practical considerations apply to clinical trials testing this compound combinations?
- Teratogenicity risks : this compound is contraindicated in pregnancy due to animal model evidence; trials require stringent contraception protocols .
- Adverse event monitoring : Severe cutaneous reactions (e.g., Stevens-Johnson syndrome) linked to sulfadoxine combinations necessitate real-time pharmacovigilance .
Retrosynthesis Analysis
AI-Powered Synthesis Planning: Our tool employs the Template_relevance Pistachio, Template_relevance Bkms_metabolic, Template_relevance Pistachio_ringbreaker, Template_relevance Reaxys, Template_relevance Reaxys_biocatalysis model, leveraging a vast database of chemical reactions to predict feasible synthetic routes.
One-Step Synthesis Focus: Specifically designed for one-step synthesis, it provides concise and direct routes for your target compounds, streamlining the synthesis process.
Accurate Predictions: Utilizing the extensive PISTACHIO, BKMS_METABOLIC, PISTACHIO_RINGBREAKER, REAXYS, REAXYS_BIOCATALYSIS database, our tool offers high-accuracy predictions, reflecting the latest in chemical research and data.
Strategy Settings
| Precursor scoring | Relevance Heuristic |
|---|---|
| Min. plausibility | 0.01 |
| Model | Template_relevance |
| Template Set | Pistachio/Bkms_metabolic/Pistachio_ringbreaker/Reaxys/Reaxys_biocatalysis |
| Top-N result to add to graph | 6 |
Feasible Synthetic Routes





Featured Recommendations
| Most viewed |
|
|
|---|---|---|
| Most popular with customers |
|
Disclaimer and Information on In-Vitro Research Products
Please be aware that all articles and product information presented on BenchChem are intended solely for informational purposes. The products available for purchase on BenchChem are specifically designed for in-vitro studies, which are conducted outside of living organisms. In-vitro studies, derived from the Latin term "in glass," involve experiments performed in controlled laboratory settings using cells or tissues. It is important to note that these products are not categorized as medicines or drugs, and they have not received approval from the FDA for the prevention, treatment, or cure of any medical condition, ailment, or disease. We must emphasize that any form of bodily introduction of these products into humans or animals is strictly prohibited by law. It is essential to adhere to these guidelines to ensure compliance with legal and ethical standards in research and experimentation.
