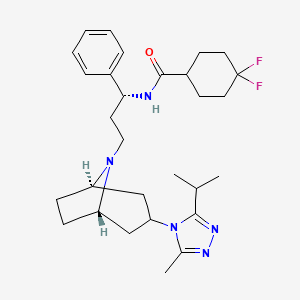
Maraviroc
Overview
Description
Maraviroc is a first-in-class CCR5 antagonist approved in 2007 for treating CCR5-tropic HIV-1 infection in treatment-experienced patients . It uniquely targets the host CCR5 receptor, a chemokine coreceptor required for viral entry, by allosterically inhibiting interactions between HIV-1 gp120 and CCR5 . Unlike most antiretrovirals (ARVs) that target viral enzymes (e.g., reverse transcriptase or protease), this compound blocks viral entry into CD4+ T-cells .
Pharmacokinetics (PK):
- This compound exhibits non-linear absorption due to saturation of P-glycoprotein (Pgp)-mediated efflux in the gut at higher doses .
- Peak plasma concentrations occur within 0.5–4 hours post-dose .
- Absolute oral bioavailability is 23% at 100 mg, increasing to 33% at 300 mg due to reduced first-pass metabolism .
- Primarily metabolized by CYP3A4, with renal clearance accounting for <25% of total elimination (increases to 70% with CYP3A4 inhibitors) .
Preparation Methods
Historical Development of Maraviroc Synthesis
Original Medicinal Synthesis by Pfizer (WO0190106)
The inaugural synthesis of this compound, disclosed in Pfizer’s 2001 patent, established a nine-step sequence beginning with tropinone (Formula II). Key stages included oxime formation via hydroxylamine in pyridine, sodium-mediated reduction to exo-amine IV, and sequential triazole cyclization. A critical bottleneck emerged in the synthesis of chiral aldehyde VIII, requiring cryogenic (−78°C) hydride reduction of tert-butyloxycarbonyl-protected β-amino ester XII to prevent over-reduction. This step’s operational complexity and low tolerance for scale-up necessitated later refinements.
Process Chemistry Optimizations (2008)
Two landmark 2008 studies in Organic Process Research & Development addressed scalability:
- Protecting Group Strategy : Replacing benzyl with carbobenzyloxy (Cbz) groups enabled room-temperature reductions, eliminating cryogenic steps.
- Oxidation Protocol : Parikh-Doering conditions (SO₃·pyridine, DMSO, Et₃N) converted alcohol XIV to aldehyde XV without epimerization, achieving 85% yield.
- Direct Amidation Attempts : Efforts to bypass aldehyde intermediates via β-amino acid coupling failed due to carbonyl reduction challenges, underscoring the need for alternative routes.
Transition Metal-Catalyzed "Borrowing Hydrogen" Alkylation
Mechanistic Basis and Catalyst Systems
The "borrowing hydrogen" method, exemplified in WO2014173375A1, leverages transition metals (e.g., Ir, Ru) to transiently dehydrogenate alcohols, forming reactive carbonyl intermediates that undergo nucleophilic attack by amines. Key advantages include:
- Avoidance of Alkyl Halides : Eliminates genotoxic byproducts inherent in traditional SN2 alkylation.
- Atom Economy : Direct use of alcohols circumvents pre-activation steps (e.g., mesylation).
For this compound, the reaction couples tropane triazole VII with (S)-amido alcohol XVII using [Cp*IrCl₂]₂ (2.5 mol%) and molecular sieves (3Å) in toluene at 120°C (Table 1).
Table 1: Catalytic Alkylation Conditions and Outcomes
| Catalyst Loading | Temperature (°C) | Time (h) | Yield (%) | Purity (%) |
|---|---|---|---|---|
| 2.5 mol% | 120 | 26 | 60 | 99.3 |
| 5.0 mol% | 110 | 18 | 83 | 97.5 |
Process Optimization and Challenges
- Solvent Selection : Toluene outperformed polar aprotic solvents (DMF, THF) by minimizing side reactions.
- Additive Effects : Molecular sieves (1.5 g/g substrate) suppressed imine hydrolysis, crucial for maintaining reaction efficiency.
- Temperature Sensitivity : Reactions below 100°C stalled at <20% conversion, while exceeding 130°C promoted decomposition.
Organocatalytic Enantioselective Synthesis of β-Amino Aldehyde
Zhao et al. (2010) pioneered an organocatalytic route to this compound’s chiral β-amino aldehyde fragment using proline-derived catalysts. This method:
- Achieved 98% enantiomeric excess (ee) via asymmetric Mannich reactions.
- Enabled gram-scale synthesis of this compound analogues for structure-activity studies.
- Reduced reliance on transition metals, appealing for pharmaceutical manufacturing.
However, this approach required post-functionalization steps (e.g., reductive amination) to assemble the full this compound scaffold, limiting its overall step economy compared to iridium-catalyzed methods.
Isolation and Purification Protocols
Acidic Salt Formation
Crude this compound (60–90% purity) was purified via hydrochloride salt formation:
- Acid Selection : HCl provided optimal crystallinity vs. HBr (hygroscopic) or citric acid (low yield).
- Basification : Adjusting to pH 12 with Na₂CO₃ precipitated free base, extracted into dichloromethane.
- Crystallization : Ethyl acetate/hexane (1:1) afforded 99.3% pure this compound with 60% recovery.
Purity-Yield Trade-offs
- Activated Carbon Treatment : Reduced colored impurities but adsorbed 5–7% product.
- Solvent Swapping : Replacing dichloromethane with ethyl acetate improved crystallization kinetics but increased processing time.
Pharmaceutical Formulation Strategies (WO2012114352A2)
Direct Compression vs. Dry Granulation
This compound’s poor compressibility necessitated innovative excipient blends:
- Disintegrant-Dispersant Synergy : Croscarmellose sodium (4%) + colloidal SiO₂ (2%) achieved <1-minute disintegration.
- Particle Size Control : d₉₀ ≤150 µm ensured content uniformity in 300 mg tablets.
Table 2: Tablet Composition and Performance
| Component | Concentration (%) | Function |
|---|---|---|
| This compound | 25 | API |
| Microcrystalline cellulose | 60 | Diluent |
| Croscarmellose Na | 4 | Disintegrant |
| Colloidal SiO₂ | 2 | Dispersant |
| Mg stearate | 1 | Lubricant |
Chemical Reactions Analysis
Maraviroc undergoes several types of chemical reactions, including:
Oxidation: this compound can be oxidized under specific conditions, leading to the formation of various oxidation products.
Reduction: Reduction reactions can modify the functional groups within this compound, altering its chemical properties.
Substitution: This compound can undergo substitution reactions where one functional group is replaced by another. Common reagents used in these reactions include oxidizing agents, reducing agents, and various catalysts.
Scientific Research Applications
Antiretroviral Therapy
Efficacy in HIV Treatment
Maraviroc is primarily indicated for treatment-experienced patients infected with R5-tropic HIV-1. Clinical trials, such as the MOTIVATE studies, have demonstrated its efficacy in reducing viral loads and improving immunological responses. In these studies, patients treated with this compound showed significantly greater reductions in HIV-1 RNA levels compared to those receiving placebo, with durable responses observed over extended follow-ups .
Long-Term Safety Profile
A five-year safety evaluation indicated that this compound was generally well-tolerated among patients, with low rates of adverse events and no significant increase in serious clinical outcomes like hepatic failure or malignancy . This safety profile supports its continued use in long-term antiretroviral therapy.
Microbicide Development
Topical Application for HIV Prevention
Recent research has explored this compound's potential as a topical microbicide. A study demonstrated that a this compound gel formulation provided complete protection against vaginal HIV-1 challenges in humanized mice models . This finding suggests that this compound could be developed as an effective microbicide to prevent sexual transmission of HIV, particularly in populations at high risk.
| Study | Model | Outcome |
|---|---|---|
| PLOS One (2011) | Humanized mice | Complete protection against vaginal HIV-1 challenge with this compound gel |
| Nature (2020) | Patient cohort | Reactivation of latent HIV with prolonged this compound administration |
Reactivation of Latent HIV
Role as a Latency-Reversing Agent
This compound has been investigated for its ability to reactivate latent HIV reservoirs. A study indicated that prolonged administration of this compound led to increased levels of viral RNA expression in patients previously on suppressive antiretroviral therapy, suggesting its potential role as a latency-reversing agent . This application is crucial for strategies aimed at achieving a functional cure for HIV.
Immunological Effects
Impact on T-cell Activation
Research has shown that this compound intensification can unexpectedly increase T-cell activation within peripheral blood and rectal mucosa during treated HIV infection. This phenomenon raises questions about the drug's effects on immune system dynamics and its potential implications for long-term treatment strategies .
Neuroprotection Research
Exploration in Neuroinflammatory Conditions
Recent studies have examined this compound's effects beyond HIV treatment, particularly regarding neuroprotection. It has been shown to reduce inflammation in vitro without exerting direct cytotoxic effects on astroglial cells, indicating potential applications in neuroinflammatory diseases . Although these findings are preliminary, they suggest a broader therapeutic scope for this compound.
Mechanism of Action
Maraviroc functions as an entry inhibitor by selectively binding to the CCR5 receptor on the surface of human cells . This binding prevents the HIV-1 gp120 protein from associating with the CCR5 receptor, thereby blocking the virus from entering the host cell . The drug acts as a negative allosteric modulator of the CCR5 receptor, inducing a conformational change that inhibits the interaction between the receptor and the virus .
Comparison with Similar Compounds
Comparison with Efavirenz (NNRTI)
Efficacy
- In the 5-year MERIT trial, maraviroc demonstrated non-inferiority to efavirenz in treatment-naive patients with CCR5-tropic HIV-1 (similar viral suppression rates: 68% vs. 63%) .
- Efavirenz, a non-nucleoside reverse transcriptase inhibitor (NNRTI), targets viral replication, while this compound prevents viral entry .
Drug-Drug Interactions (DDIs)
- Efavirenz, a potent CYP3A4 inducer, reduces this compound exposure by 45%, necessitating a dose increase from 300 mg to 600 mg twice daily .
Comparison with Raltegravir (Integrase Inhibitor)
- Efficacy: In the STARTMRK trial, raltegravir (integrase inhibitor) demonstrated comparable efficacy to efavirenz (83% vs. 84% viral suppression at 48 weeks), similar to this compound .
- Mechanism: Raltegravir inhibits viral DNA integration, while this compound blocks entry. This compound requires tropism testing (CCR5-tropic virus), whereas raltegravir is tropism-independent .
Pharmacokinetic Comparison with Other CYP3A4-Substrate ARVs
Structural and Mechanistic Uniqueness
This compound binds to an allosteric site on CCR5, distinct from the chemokine (e.g., CCL3/5) and gp120 binding pockets, preventing conformational changes required for viral fusion . In contrast, CXCR4 antagonists (e.g., AMD3100) target a different receptor, highlighting this compound’s specificity for CCR5-tropic HIV-1 .
Biological Activity
Maraviroc, also known as UK-427857, is a selective antagonist of the CCR5 receptor, which plays a crucial role in the entry of HIV-1 into host cells. This compound represents the first drug in a new class of antiretroviral agents that target host proteins rather than viral components. Its unique mechanism of action and pharmacological properties have made it a significant focus of research in the treatment of HIV-1 infections.
This compound inhibits HIV-1 entry by binding to the CCR5 receptor on the surface of CD4+ T cells. This binding prevents the viral envelope protein gp120 from interacting with CCR5, effectively blocking the fusion process necessary for viral entry into host cells. The geometric mean 90% inhibitory concentration (IC90) for this compound against various CCR5-tropic HIV-1 strains is approximately 2.0 nM, demonstrating its potent antiviral activity against a wide range of viral isolates .
In Vitro Studies
In vitro studies have shown that this compound exhibits:
- Potent Anti-HIV Activity : Effective against 200 clinically derived HIV-1 envelope-recombinant pseudoviruses, including those resistant to other drug classes.
- Selectivity for CCR5 : this compound has been confirmed to be highly selective for the CCR5 receptor, with no significant effects on other receptors or enzymes, including hERG ion channels, indicating a favorable safety profile .
Pharmacokinetics
This compound is characterized by its oral bioavailability and pharmacokinetic properties that support once or twice daily dosing. Preclinical models predict human pharmacokinetics consistent with these dosing regimens, allowing for convenient administration in clinical settings .
Clinical Efficacy
Clinical trials have demonstrated that this compound effectively reduces viral load in patients with CCR5-tropic HIV-1. It has been shown to improve immune function and reduce the risk of disease progression in treated individuals. The drug's efficacy is further supported by studies highlighting its ability to maintain viral suppression over extended periods .
Table 1: Summary of this compound's Biological Activity
| Property | Value |
|---|---|
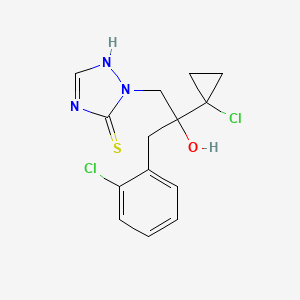
| Chemical Name | 4,4-Difluoro-N-{(1S)-3-[3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1-phenylpropyl}cyclohexanecarboxamide |
| IC90 Against CCR5-tropic HIV-1 | 2.0 nM |
| Selectivity Index (hERG) | >10 μM |
| Oral Bioavailability | Yes |
| Recommended Dosing | Once or twice daily |
Table 2: Clinical Trial Results for this compound
| Study Phase | Participants | Outcome |
|---|---|---|
| Phase II | 200 | Significant reduction in viral load |
| Phase III | 500 | Improved CD4+ T cell counts |
| Long-term follow-up | 300 | Sustained viral suppression |
Case Study 1: Efficacy in Treatment-Experienced Patients
A study involving treatment-experienced patients showed that those receiving this compound as part of their regimen experienced a significant decline in plasma HIV RNA levels compared to those on standard therapy alone. This highlights this compound's potential as an effective option for patients with limited treatment choices due to resistance .
Case Study 2: Safety Profile Assessment
In a long-term safety assessment involving over 1000 participants, this compound demonstrated a favorable safety profile with minimal adverse effects reported. The most common side effects were mild and included headaches and gastrointestinal disturbances. Importantly, no significant cardiovascular events were noted during the trial period .
Properties
IUPAC Name |
4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-phenylpropyl]cyclohexane-1-carboxamide |
Source


|
|---|---|---|
| Details | Computed by Lexichem TK 2.7.0 (PubChem release 2021.05.07) | |
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
InChI |
InChI=1S/C29H41F2N5O/c1-19(2)27-34-33-20(3)36(27)25-17-23-9-10-24(18-25)35(23)16-13-26(21-7-5-4-6-8-21)32-28(37)22-11-14-29(30,31)15-12-22/h4-8,19,22-26H,9-18H2,1-3H3,(H,32,37) |
Source
|
| Details | Computed by InChI 1.0.6 (PubChem release 2021.05.07) | |
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
InChI Key |
GSNHKUDZZFZSJB-UHFFFAOYSA-N |
Source
|
| Details | Computed by InChI 1.0.6 (PubChem release 2021.05.07) | |
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Canonical SMILES |
CC1=NN=C(N1C2CC3CCC(C2)N3CCC(C4=CC=CC=C4)NC(=O)C5CCC(CC5)(F)F)C(C)C |
Source
|
| Details | Computed by OEChem 2.3.0 (PubChem release 2021.05.07) | |
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Molecular Formula |
C29H41F2N5O |
Source
|
| Details | Computed by PubChem 2.1 (PubChem release 2021.05.07) | |
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Molecular Weight |
513.7 g/mol |
Source
|
| Details | Computed by PubChem 2.1 (PubChem release 2021.05.07) | |
| Source | PubChem | |
| URL | https://pubchem.ncbi.nlm.nih.gov | |
| Description | Data deposited in or computed by PubChem | |
Retrosynthesis Analysis
AI-Powered Synthesis Planning: Our tool employs the Template_relevance Pistachio, Template_relevance Bkms_metabolic, Template_relevance Pistachio_ringbreaker, Template_relevance Reaxys, Template_relevance Reaxys_biocatalysis model, leveraging a vast database of chemical reactions to predict feasible synthetic routes.
One-Step Synthesis Focus: Specifically designed for one-step synthesis, it provides concise and direct routes for your target compounds, streamlining the synthesis process.
Accurate Predictions: Utilizing the extensive PISTACHIO, BKMS_METABOLIC, PISTACHIO_RINGBREAKER, REAXYS, REAXYS_BIOCATALYSIS database, our tool offers high-accuracy predictions, reflecting the latest in chemical research and data.
Strategy Settings
| Precursor scoring | Relevance Heuristic |
|---|---|
| Min. plausibility | 0.01 |
| Model | Template_relevance |
| Template Set | Pistachio/Bkms_metabolic/Pistachio_ringbreaker/Reaxys/Reaxys_biocatalysis |
| Top-N result to add to graph | 6 |
Feasible Synthetic Routes









Disclaimer and Information on In-Vitro Research Products
Please be aware that all articles and product information presented on BenchChem are intended solely for informational purposes. The products available for purchase on BenchChem are specifically designed for in-vitro studies, which are conducted outside of living organisms. In-vitro studies, derived from the Latin term "in glass," involve experiments performed in controlled laboratory settings using cells or tissues. It is important to note that these products are not categorized as medicines or drugs, and they have not received approval from the FDA for the prevention, treatment, or cure of any medical condition, ailment, or disease. We must emphasize that any form of bodily introduction of these products into humans or animals is strictly prohibited by law. It is essential to adhere to these guidelines to ensure compliance with legal and ethical standards in research and experimentation.