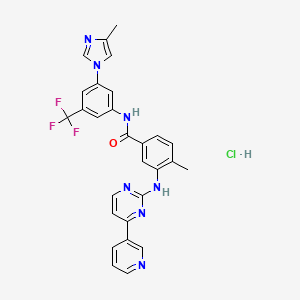
Nilotinib hydrochloride
Cat. No. B1258797
Key on ui cas rn:
923288-95-3
M. Wt: 566.0 g/mol
InChI Key: VTGGYCCJUPYZSX-UHFFFAOYSA-N
Attention: For research use only. Not for human or veterinary use.
Patent
US08829015B2
Procedure details


4.0 g 4-methyl-N-[3-(4-methyl-imidazol-1-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-benzamide free base is dissolved in 60 mL methanol at 50° C. 1.05 equivalent (688.7 μL) of hydrochloric acid is added as a solution in 2 mL of methanol. The solution is let stand for 60 minutes at 50° C. The solution is cooled down to 42° C. and kept at this temperature for 15 minutes. A suspension of 4 mg of 4-methyl-N-[3-(4-methyl-imidazol-1-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-benzamide hydrochloride in methanol (40 mg)/water (0.4 mg) homogenized for 10 seconds in an ultrasonic bath is added. The suspension is let stand for 2.5 hours at 42° C., then cooled down in 7 hours at 20° C. The suspension remains at 20° C. for 56 hours. The suspension is not filtrated before analysis. The dimethanol solvate form SB of the hydrochloride salt of 4-methyl-N-[3-(4-methyl-imidazol-1-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-benzamide is obtained.
Quantity
4 g
Type
reactant
Reaction Step One

Quantity
4 mg
Type
reactant
Reaction Step Three

Name
Identifiers
|
REACTION_CXSMILES
|
CC1C=C[C:5]([C:6](NC2C=C(C(F)(F)F)C=C(N3C=C(C)N=C3)C=2)=[O:7])=CC=1NC1N=C(C2C=NC=CC=2)C=CN=1.Cl.Cl.[CH3:42][C:43]1[CH:67]=[CH:66][C:46]([C:47]([NH:49][C:50]2[CH:55]=[C:54]([C:56]([F:59])([F:58])[F:57])[CH:53]=[C:52]([N:60]3[CH:64]=[C:63]([CH3:65])[N:62]=[CH:61]3)[CH:51]=2)=[O:48])=[CH:45][C:44]=1[NH:68][C:69]1[N:74]=[C:73]([C:75]2[CH:76]=[N:77][CH:78]=[CH:79][CH:80]=2)[CH:72]=[CH:71][N:70]=1.[OH2:81].[CH3:82][OH:83]>>[CH2:6]([O:7][CH2:47][OH:48])[CH2:5][O:81][CH2:82][OH:83].[CH3:42][C:43]1[CH:67]=[CH:66][C:46]([C:47]([NH:49][C:50]2[CH:55]=[C:54]([C:56]([F:57])([F:58])[F:59])[CH:53]=[C:52]([N:60]3[CH:64]=[C:63]([CH3:65])[N:62]=[CH:61]3)[CH:51]=2)=[O:48])=[CH:45][C:44]=1[NH:68][C:69]1[N:74]=[C:73]([C:75]2[CH:76]=[N:77][CH:78]=[CH:79][CH:80]=2)[CH:72]=[CH:71][N:70]=1 |f:2.3|
|
Inputs
Step One
|
Name
|
|
|
Quantity
|
4 g
|
|
Type
|
reactant
|
|
Smiles
|
CC1=C(C=C(C(=O)NC2=CC(=CC(=C2)C(F)(F)F)N2C=NC(=C2)C)C=C1)NC1=NC=CC(=N1)C=1C=NC=CC1
|
|
Name
|
|
|
Quantity
|
60 mL
|
|
Type
|
reactant
|
|
Smiles
|
CO
|
Step Two
|
Name
|
|
|
Quantity
|
688.7 μL
|
|
Type
|
reactant
|
|
Smiles
|
Cl
|
|
Name
|
|
|
Quantity
|
2 mL
|
|
Type
|
reactant
|
|
Smiles
|
CO
|
Step Three
|
Name
|
|
|
Quantity
|
4 mg
|
|
Type
|
reactant
|
|
Smiles
|
Cl.CC1=C(C=C(C(=O)NC2=CC(=CC(=C2)C(F)(F)F)N2C=NC(=C2)C)C=C1)NC1=NC=CC(=N1)C=1C=NC=CC1
|
|
Name
|
|
|
Quantity
|
0.4 mg
|
|
Type
|
reactant
|
|
Smiles
|
O
|
|
Name
|
|
|
Quantity
|
40 mg
|
|
Type
|
reactant
|
|
Smiles
|
CO
|
Conditions
Temperature
|
Control Type
|
UNSPECIFIED
|
|
Setpoint
|
42 °C
|
Stirring
|
Type
|
CUSTOM
|
|
Details
|
homogenized for 10 seconds in an ultrasonic bath
|
|
Rate
|
UNSPECIFIED
|
|
RPM
|
0
|
Other
|
Conditions are dynamic
|
1
|
|
Details
|
See reaction.notes.procedure_details.
|
Workups
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
for 60 minutes
|
|
Duration
|
60 min
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
at 50° C
|
ADDITION
|
Type
|
ADDITION
|
|
Details
|
is added
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
for 2.5 hours
|
|
Duration
|
2.5 h
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
at 42° C.
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
cooled down in 7 hours at 20° C
|
|
Duration
|
7 h
|
WAIT
|
Type
|
WAIT
|
|
Details
|
The suspension remains at 20° C. for 56 hours
|
|
Duration
|
56 h
|
FILTRATION
|
Type
|
FILTRATION
|
|
Details
|
The suspension is not filtrated before analysis
|
Outcomes
Product
Details
Reaction Time |
15 min |
|
Name
|
|
|
Type
|
product
|
|
Smiles
|
C(COCO)OCO
|
|
Name
|
|
|
Type
|
product
|
|
Smiles
|
|
|
Name
|
|
|
Type
|
product
|
|
Smiles
|
CC1=C(C=C(C(=O)NC2=CC(=CC(=C2)C(F)(F)F)N2C=NC(=C2)C)C=C1)NC1=NC=CC(=N1)C=1C=NC=CC1
|
Source
|
Source
|
Open Reaction Database (ORD) |
|
Description
|
The Open Reaction Database (ORD) is an open-access schema and infrastructure for structuring and sharing organic reaction data, including a centralized data repository. The ORD schema supports conventional and emerging technologies, from benchtop reactions to automated high-throughput experiments and flow chemistry. Our vision is that a consistent data representation and infrastructure to support data sharing will enable downstream applications that will greatly improve the state of the art with respect to computer-aided synthesis planning, reaction prediction, and other predictive chemistry tasks. |
Patent
US08829015B2
Procedure details
4.0 g 4-methyl-N-[3-(4-methyl-imidazol-1-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-benzamide free base is dissolved in 60 mL methanol at 50° C. 1.05 equivalent (688.7 μL) of hydrochloric acid is added as a solution in 2 mL of methanol. The solution is let stand for 60 minutes at 50° C. The solution is cooled down to 42° C. and kept at this temperature for 15 minutes. A suspension of 4 mg of 4-methyl-N-[3-(4-methyl-imidazol-1-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-benzamide hydrochloride in methanol (40 mg)/water (0.4 mg) homogenized for 10 seconds in an ultrasonic bath is added. The suspension is let stand for 2.5 hours at 42° C., then cooled down in 7 hours at 20° C. The suspension remains at 20° C. for 56 hours. The suspension is not filtrated before analysis. The dimethanol solvate form SB of the hydrochloride salt of 4-methyl-N-[3-(4-methyl-imidazol-1-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-benzamide is obtained.
Quantity
4 g
Type
reactant
Reaction Step One
Quantity
4 mg
Type
reactant
Reaction Step Three
Name
Identifiers
|
REACTION_CXSMILES
|
CC1C=C[C:5]([C:6](NC2C=C(C(F)(F)F)C=C(N3C=C(C)N=C3)C=2)=[O:7])=CC=1NC1N=C(C2C=NC=CC=2)C=CN=1.Cl.Cl.[CH3:42][C:43]1[CH:67]=[CH:66][C:46]([C:47]([NH:49][C:50]2[CH:55]=[C:54]([C:56]([F:59])([F:58])[F:57])[CH:53]=[C:52]([N:60]3[CH:64]=[C:63]([CH3:65])[N:62]=[CH:61]3)[CH:51]=2)=[O:48])=[CH:45][C:44]=1[NH:68][C:69]1[N:74]=[C:73]([C:75]2[CH:76]=[N:77][CH:78]=[CH:79][CH:80]=2)[CH:72]=[CH:71][N:70]=1.[OH2:81].[CH3:82][OH:83]>>[CH2:6]([O:7][CH2:47][OH:48])[CH2:5][O:81][CH2:82][OH:83].[CH3:42][C:43]1[CH:67]=[CH:66][C:46]([C:47]([NH:49][C:50]2[CH:55]=[C:54]([C:56]([F:57])([F:58])[F:59])[CH:53]=[C:52]([N:60]3[CH:64]=[C:63]([CH3:65])[N:62]=[CH:61]3)[CH:51]=2)=[O:48])=[CH:45][C:44]=1[NH:68][C:69]1[N:74]=[C:73]([C:75]2[CH:76]=[N:77][CH:78]=[CH:79][CH:80]=2)[CH:72]=[CH:71][N:70]=1 |f:2.3|
|
Inputs
Step One
|
Name
|
|
|
Quantity
|
4 g
|
|
Type
|
reactant
|
|
Smiles
|
CC1=C(C=C(C(=O)NC2=CC(=CC(=C2)C(F)(F)F)N2C=NC(=C2)C)C=C1)NC1=NC=CC(=N1)C=1C=NC=CC1
|
|
Name
|
|
|
Quantity
|
60 mL
|
|
Type
|
reactant
|
|
Smiles
|
CO
|
Step Two
|
Name
|
|
|
Quantity
|
688.7 μL
|
|
Type
|
reactant
|
|
Smiles
|
Cl
|
|
Name
|
|
|
Quantity
|
2 mL
|
|
Type
|
reactant
|
|
Smiles
|
CO
|
Step Three
|
Name
|
|
|
Quantity
|
4 mg
|
|
Type
|
reactant
|
|
Smiles
|
Cl.CC1=C(C=C(C(=O)NC2=CC(=CC(=C2)C(F)(F)F)N2C=NC(=C2)C)C=C1)NC1=NC=CC(=N1)C=1C=NC=CC1
|
|
Name
|
|
|
Quantity
|
0.4 mg
|
|
Type
|
reactant
|
|
Smiles
|
O
|
|
Name
|
|
|
Quantity
|
40 mg
|
|
Type
|
reactant
|
|
Smiles
|
CO
|
Conditions
Temperature
|
Control Type
|
UNSPECIFIED
|
|
Setpoint
|
42 °C
|
Stirring
|
Type
|
CUSTOM
|
|
Details
|
homogenized for 10 seconds in an ultrasonic bath
|
|
Rate
|
UNSPECIFIED
|
|
RPM
|
0
|
Other
|
Conditions are dynamic
|
1
|
|
Details
|
See reaction.notes.procedure_details.
|
Workups
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
for 60 minutes
|
|
Duration
|
60 min
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
at 50° C
|
ADDITION
|
Type
|
ADDITION
|
|
Details
|
is added
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
for 2.5 hours
|
|
Duration
|
2.5 h
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
at 42° C.
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
cooled down in 7 hours at 20° C
|
|
Duration
|
7 h
|
WAIT
|
Type
|
WAIT
|
|
Details
|
The suspension remains at 20° C. for 56 hours
|
|
Duration
|
56 h
|
FILTRATION
|
Type
|
FILTRATION
|
|
Details
|
The suspension is not filtrated before analysis
|
Outcomes
Product
Details
Reaction Time |
15 min |
|
Name
|
|
|
Type
|
product
|
|
Smiles
|
C(COCO)OCO
|
|
Name
|
|
|
Type
|
product
|
|
Smiles
|
|
|
Name
|
|
|
Type
|
product
|
|
Smiles
|
CC1=C(C=C(C(=O)NC2=CC(=CC(=C2)C(F)(F)F)N2C=NC(=C2)C)C=C1)NC1=NC=CC(=N1)C=1C=NC=CC1
|
Source
|
Source
|
Open Reaction Database (ORD) |
|
Description
|
The Open Reaction Database (ORD) is an open-access schema and infrastructure for structuring and sharing organic reaction data, including a centralized data repository. The ORD schema supports conventional and emerging technologies, from benchtop reactions to automated high-throughput experiments and flow chemistry. Our vision is that a consistent data representation and infrastructure to support data sharing will enable downstream applications that will greatly improve the state of the art with respect to computer-aided synthesis planning, reaction prediction, and other predictive chemistry tasks. |
